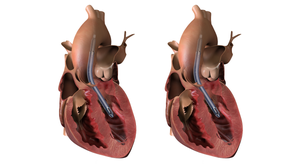
Beating Heart
CardiovascularBrioHealth Score IDE Approval for BioVAD System Trial EnrollmentBrioHealth Score IDE Approval for BioVAD System Trial Enrollment
In the study, the device will be evaluated relative to technology that has already been FDA approved.











 designed to help the heart pump blood to the rest of the body.")
















.png?width=300&auto=webp&quality=80&disable=upscale "plastic syringe")


.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "EU MDR")
.png?width=300&auto=webp&quality=80&disable=upscale "Axonics R20 Rechargeable Sacral Neuromodulation System")


.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")
Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.