Sign up for the QMED & MD+DI Daily newsletter.
FDA Guidance for ISO 10993-1: What to Expect
Several noteworthy changes are anticipated in a new FDA guidance document on biological evaluation and biocompatibility testing of medical devices due out sometime this year.
January 20, 2016
5 Min Read

Several noteworthy changes are anticipated in a new FDA guidance document on biological evaluation and biocompatibility testing of medical devices due out sometime this year.
Audrey Turley
There has been a lot of chatter over the past two years about FDA’s draft guidance on ISO 10993. This chatter is now turning into action, as can be seen in the feedback medical device manufacturers are receiving from FDA through their submissions. A final version of this guidance was expected in December 2015 and is now anticipated during the first quarter of 2016.
The delay is most likely due to heavy discussions at the ISO meetings held in Lund, Sweden, in June 2015. Throughout these meetings regulatory bodies, medical device manufacturers, and laboratory experts from around the world weighed in on the ISO 10993 standards. Intense conversations ensued, trying to clarify the interpretation of the literature and standards as they relate to the shift from animal testing to a risk-based approach using material and/or chemical characterization.
Based on these conversations, direct feedback from FDA, and recently published research, we anticipate a few areas of testing to be addressed differently in the upcoming guidance document. These include genotoxicity, hemocompatibility, pyrogenicity and changes to the ISO 10993 table.
Genotoxicity
FDA specifically states that it “…traditionally request[s] genotoxicity testing, even if the device will not have permanent duration of use.” Due to some recent research by Robert Przygoda, principal scientist at Johnson & Johnson, and colleagues, much discussion has been held regarding the applicability of the current genotoxicity procedures to medical devices.
The concern is that most of the genotoxicity methods have been adapted from chemical protocols and do not transfer well to medical device extract testing. Medical devices need to be extracted, and the extraction fluid is then applied to the genotoxicity test system. A small amount of this extract (0.5–10%) is all that can be added to the test system in order to maintain the nutrient balance needed to obtain viable results. The test system loses sensitivity due to this dilution.
The genotoxic potential of a medical device can be assessed through an alternate path involving a risk based approach. This can be performed by identifying hazardous compounds or processes used in a medical device through chemical characterization testing. These hazards can then be researched for their genotoxic potential through published literature and history of use. It is anticipated that genotoxicity testing will still be listed as a required endpoint; however, alternate options that do not involve specific genotoxicity testing will be preferred.
Hemocompatibility
The in vivo canine venous test model to assess thrombogenicity is under review for alternative in vitro methods. The in vivo model has significant flaws, and many devices cannot be placed properly in the test subject for an accurate detection of thrombus potential. Encouragement to perform in vitro hemolysis assays and to incorporate thrombosis in functionality testing is expected.
Currently, FDA is asking for three human donors to be used in all hemolysis tests. There has been much debate regarding whether human blood can be pooled for in vitro hemolysis assays. The concern is that agglutination, the clumping of red blood cells due to antibody interactions, will occur. This could lead to a false positive response in testing. Guidance as to how blood may be pooled (after blood typing and Rh factor identification) is expected, outlining which blood groups are compatible.
Pyrogenicity
It is anticipated that bacterial endotoxin testing will be required for all implants especially orthopedics. In the past, this testing was recommended based on sterility testing requirements and labeling as “nonpyrogenic.” However, pyrogenicity is another way to screen a device for cleanliness. The Limulus amebocyte lysate (LAL) assay is currently used to screen for pyrogenicity, this assay specifically looks for endotoxins. Endotoxins are found in Gram negative bacteria which can commonly arise from water processing systems. FDA suggests screening for bacterial endotoxin using the LAL assay to demonstrate cleanliness in the manufacturing process.
Initial Evaluation Tests for Consideration (Table 1 in Attachment A of the draft)
The industry is moving towards a risk based approach to biocompatibility, beginning with the materials used in medical devices. The current draft guidance for biological evaluation of medical devices issued April 23, 2013 discusses a big picture approach to biocompatibility assessment of medical devices. The biggest change in the final FDA guidance document is expected to occur to the chart in Attachment A – Table 1. We anticipate an additional column in the table adding “Material/Chemical Characterization”. Material/Chemical Characterization and Cytotoxicity testing would support a risk based beginning to determine how the other test categories need to be addressed; through testing or risk analysis.
(click to enlarge)
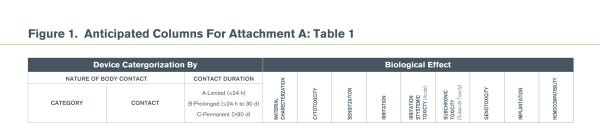
(click to enlarge)

Conclusion
ISO 10993 should not be considered a checklist to be completed for biological safety evaluations. The intent of the standard is to establish a toxicological profile of the medical device. FDA’s draft guidance already contains specific language to support a risk-based approach. This direction is not anticipated to change; in fact, it is expected that further language and action to support a risk-based approach is likely as the industry strives to perform scientific evaluations of medical devices that benefit all, including a reduction in the use of test animals.
Audrey Turley is a research scientist at Nelson Laboratories Inc. Reach her at [email protected]. See Nelson Laboratories at MD&M West (Booth #3131) and hear Turley’s presentation on a new approach to assessing biocompatibility for medical devices in the Tech Theater on February 9 at 12:30 p.m.
Correction: Based on industry feedback, the full image in Figure 1 that appeared in an earlier version of this article was replaced with a top view of the table. Figure 2 was also added to provide an outline for how medical device manufacturers may approach biocompatibility.
[image courtesy of SUWIT RITJAROON/FREEDIGITALPHOTOS.NET]
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
