Sign up for the QMED & MD+DI Daily newsletter.
Best Practices: Managing a CAPA System
Barbara K. Immel
June 1, 2006
17 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Best Practices: Managing a CAPA System")
Establishing a corrective and preventive action (CAPA) system is a quality system regulation (QSR) requirement for medical device firms marketing products in the United States. Although FDA has always expected regulated industries to perform corrective and preventive actions, establishing a unified CAPA system also makes good business sense. The sidebar, “The One-Minute CAPA,” on page 50 gives a quick overview of important practices in a CAPA system.
The first and most important step in creating any system is to include all regulatory requirements in the firm's standard operating procedures (SOPs).
When the device CGMPs were revised and incorporated into the QSR in 1996, FDA added an entire section on CAPA. FDA considers it to be a major subsystem of a manufacturer's quality system.1 The following seven key elements from 21 CFR 820.100 should be included in a firm's SOPs:
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
Definitions
In ISO 9000 standards, corrective action is defined as “action to eliminate the cause of a detected nonconformity or other undesirable situation.” There can be more than one cause for a nonconformity. Corrective action is taken to prevent recurrence, whereas preventive action is taken to prevent occurrence. There is also a slight distinction between correction and corrective action. A correction is an “action to eliminate a detected nonconformity.” So, a correction can be, for example, rework or regrade. Preventive action is defined as “action to eliminate the cause of a potential nonconformity or other undesirable potential situation.” As with corrective action, there can be more than one cause for a potential nonconformity. Preventive action is taken to prevent an occurrence, whereas corrective action is taken to prevent recurrence.2
Corrective action is used to correct a problem, and preventive action is used to prevent the problem or potential problem from occurring. Because the QSR does not define these terms, there has been some blurring of their meaning. Therefore, it is important to define terms in the company's SOPs. Firms should also clearly define these terms for employees and explain the company's expectations in training classes.
Invest in Necessary Resources
All QSR requirements mandate that companies have staff with the training, knowledge, and experience for the job. This staffing includes experienced quality assurance (QA) staff who will manage the CAPA system. In addition, firms must invest in the databases and computer software that can perform the necessary processes. CAPA is not a paper-shuffling or document-control exercise. Some analyses of products, processes, or systems may require an R&D, manufacturing, engineering, or technical background—or a clinician's or microbiologist's review.
Some managers find that the only way to obtain additional staff and computer systems is to track department statistics, including turnaround times, number of requests, and number of rush requests. Reporting these statistics and the risks associated with not promptly addressing nonconformities is often a good way to convince senior management to provide needed resources. Device companies must include CAPA information in their required management reviews. Sometimes the findings from an FDA or external audit may help get necessary resources as well.
Identify Data Sources
Sidebar: The One-Minute CAPA: Key Practices |
Proper data sources are critical to a CAPA system. Companies should identify the data sources they will use to identify existing problems as well as potential problems. An AdvaMed document, “Points to Consider When Preparing for an FDA Inspection Under the QSIT Corrective and Preventive Action Subsystem,” lists a number of possible sources of useful data.3
The AdvaMed guide lists a number of good data sources for identifying existing problems. Among them are acceptance activities, complaints, FDA-483 observations or warning letters, nonconformances or deviations, process monitoring data, and calibration and preventive maintenance records. The guide also recommends possible sources of data for identifying potential problems. These sources include but are not limited to acceptance activities; process monitoring data; calibration and preventive maintenance records; internal, external, supplier, or third-party audits; and customer or employee feedback.
Analyze Data Regularly
Data should be examined on a regular basis. At the very least, companies should use a suitable scientific method of analysis. Trending should be done at least quarterly to be useful. In many cases, trending should be done more frequently, such as on a monthly or daily basis.
Area supervisors and managers should be asked to routinely trend the performance of their department and to act on the results. Reviewing trends and brainstorming possible improvements with employees is an excellent way to manage a department. An added benefit is that employees are informed about nonconformities or device defects that may occur from the improper performance of their specific job, which is a QSR requirement.1
Document Procedures
It is important to define in a company's SOPs which items should be elevated to a CAPA request and who will make the final call. The final call on whether an official CAPA request should be opened should be made by an experienced QA professional.
A firm's SOPs and processes should be carefully planned. All critical CAPA items resulting from important investigations—or from continuous improvement projects—should be tracked in a centralized system. Do not try to hide information from FDA in internal audit reports, supplier audit reports, or management reviews. The agency has the right to view CAPA activities arising from such sources.
|
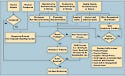
Figure 1. This flowchart shows how companies can integrate risk management into a CAPA process. Other data points (a) can include finished goods returned or credit restock. The relationship between risk management and CAPA processes depends on the output of the investigation. That process can be iterative. Source: Global Harmonization Task Force. |
In addition, it is important to remember preventive action. Some firms deal only with known problems rather than identifying potential problems and taking action before they occur. CAPA action should not only correct an immediate problem or issue; it should also prevent it or something similar from occurring or repeating.
Preventive actions may include working with a team to research, identify, and recommend possible improvements in a change control, document change request, or engineering change order system. Doing so may also include specifying, purchasing, installing, upgrading, and validating software. Figure 1, which is taken from the Global Harmonization Task Force's “Implementation of Risk Management Principles and Activities within a Quality Management System,” shows a sample flowchart illustrating process flow.4
Timeliness and Accuracy Are Key
Companies must ensure the timeliness and accuracy of data being received into their systems. Data should be received and reviewed promptly (if large volumes of data are amassed, a computer system may help capture and promptly analyze them). Data should be entered into the computer system in a timely fashion. In addition, companies must ensure that all the necessary data are being captured. Electronic systems must be 21 CFR Part 11 compliant such that records cannot be overwritten or deleted without a trace. Employees should be trained to immediately notify the QA or QS department when they deviate from an established procedure, work instruction, or batch record. In addition, firms must ensure that QA is notified immediately about major issues. A surveillance system is only as good as the information it receives.
Use Statistics
Companies must make sure that the statistical methods they use to analyze data are appropriate, and they should compare data across different sources. FDA knows that companies sometimes misuse statistics. The preamble of the QSR states: “FDA has seen far too often the misuse of statistics by manufacturers in an effort to minimize instead of address [a] problem.”1 Reading the preamble of the QSR (particularly comments 158–166) can help firms gain a better understanding of FDA's expectations. In essence, companies need to take a more global approach to analyzing their quality data. They should look across data, products, and facilities to detect recurring product problems as well as problems within their quality system.
Root-Cause Analysis Training
It is essential that employees be trained in root-cause analysis and how to conduct a problem investigation. The book Apollo Root Cause Analysis states that there are always at least two causes of any problem.5 There is always a preexisting condition and an action (or catalyst) that when combined result in a problem. Therefore, employees should always look for at least two causes of any problem. Causes often extend beyond the area or function where they are detected, so for major problems, assemble a cross-functional team to perform the root-cause analysis.
When teaching employees how to conduct a problem investigation, train them to always include a good problem definition statement. It is helpful to review pertinent FDA inspection guides during training and discuss how to investigate a problem using case studies from recent FDA-483s or warning letters.6 FDA warning letters and frequently requested FDA 483s are available on the agency's Web site.
When presenting the warning letters or 483s, ask employees to pick an interesting observation and determine how they would investigate the problem. Then have them brainstorm possible root causes for the observation and put together a recommended CAPA plan. In other words, they should practice performing a problem investigation. The sidebar above, “Employee Training: 20 Tips for Problem Investigation,” provides helpful guidance. The trainees should be taught that the depth and intensity of any investigation should match the possible risk to patients. When they suggest a CAPA item, they should also suggest how to measure its effectiveness.
Internal Audits
FDA considers audits one of the most important QA tools, so firms should routinely perform internal and quality audits. It is important to not rely only on external, FDA, or client audits—companies should also perform their own audits to ensure their CAPA systems are adequate. Current industry practice is to audit each QSR area at least once a year, and more frequently if there are problems in a particular area. To do this, sufficient resources are required. Checking the status of CAPA items (to make sure that they are completed, and that the same problems are not repeating) is an important step to take in an internal audit.
Handling Investigations and Taking Action
It should be a companywide goal to ensure that the depth and intensity of investigations match the significance of the problem. Any problem involving distributed products must be investigated with the utmost urgency, because it may require the firm to issue a recall or notify FDA through a medical device report (MDR), field alert, or biological product deviation report. The thoroughness of the CAPA recommended should also be commensurate with the possible risk to patients.
Firms should be doing a risk classification of their investigations. Also important is trending health hazard evaluation ratings from physicians, patient incidents by product, the type of incidents being reported, any corrections and FDA notifications, the reasons for product defects, etc. In the device QSR preamble, FDA states its expectation:1
…nonconforming product discovered before or after distribution [must] be investigated to the degree commensurate with the significance and risk of the nonconformity. At times, a very in-depth investigation will be necessary, while at other times a simple investigation followed by trend analysis or other appropriate tools will be acceptable… The objective…is to correct and prevent poor practices, not simply bad product. Correction and prevention of unacceptable quality system practices should result in fewer nonconformities related to product… [and fewer] problems within the quality system itself. For example, it should identify and correct improper personnel training, the failure to follow procedures, and inadequate procedures, among other things.
However, a CAPA system will not help a firm if its executives do not act promptly and appropriately. The safety of patients must always be the primary concern. In one case, a company paid $92.4 million in fines for failing to file all required MDRs for a device delivery system and implant that got stuck in patients' bodies, resulting in 12 deaths and 57 emergency surgeries. Although the company filed 172 MDRs, it failed to file 2628 additional MDRs, and it did not notify FDA about the deaths or emergency operations. Since senior management would neither act nor allow employees to file the necessary reports, seven employees of the company wrote to FDA anonymously.7
FDA states in the device QSR preamble, “any death, even if the manufacturer attributes it to user error, will be considered relevant by FDA and will have a high risk potentially associated with it.”1 It is both an ethical and a legal responsibility to report such issues to FDA rapidly. Manufacturers cannot allow harm to come to patients if they know that it can be prevented.
It's also very important to communicate known risks to patients quickly. In a recent warning letter sent to a manufacturer of implantable cardioverter-defibrillators and pacemakers, FDA cited the company for not identifying “adequate and timely actions…for correction and prevention” of the issue. The letter went on to cite the company for not informing users of the devices' potential to fail.8
Document and Implement CAPA
A clear and documented plan that includes deliverables and due dates should be developed as soon as the root causes of a problem are known. Ideally, this would be within 30 days of the discovery of the problem, unless it concerns a distributed product, in which case the plan must be developed immediately. The plan should describe exactly what the company will do when a complaint is received as well as who will do it. Some companies ask all potential team members to sign and date their commitment to complete a CAPA item within a defined time.
CAPA requests that have been open for more than a few months should require a written or periodic review to evaluate their status. Document when they are expected to be complete, why they are still open, whether additional resources are needed, etc. For long-term CAPAs, written status reports should be required every 30 days.
|
According to 21 CFR 800.100, seven steps are key to a CAPA SOP. |
The time it takes to close a corrective action item should not be a finite number in SOPs. Instead, it will often depend on the corrective action plan. If part of the plan requires purchasing equipment (plus installation, training, and validation), the due date depends on when the various action items can be completed. Each corrective action plan should define the projected completion date. Any CAPA items still outstanding after a certain time period should require a formal, documented review by QA and management to determine why they are still open. However, a CAPA request should not be closed until all items in its action plan are implemented. Also, companies should not allow scope creep. If the scope of a corrective action report or CAPA request keeps expanding, consider opening another CAPA request to track the additional work.
Companies should evaluate, verify, and validate the CAPA system before it is used. And, after it is implemented, companies should evaluate its effectiveness. Use an approved protocol to test or evaluate any major proposed actions before taking them. Solutions should prevent recurrence of the problem at all locations, not cause other unacceptable problems. They should be within the firm's control to enact, and they should provide good value for their cost.5 Consulting with the regulatory affairs department regarding any proposed changes that may require FDA approval before implementation is wise. Also, it is important to check the process in which the problem was first discovered to see whether it is repeating. Checking trends in the nonconforming process can determine whether the problem or issue has been resolved.
If a problem affects a product, firms must prevent any nonconforming product from being distributed. This requires manufacturers to truly have control over their systems. Firms need systems in place that enable them to identify nonconforming product and take immediate action. Someone must physically segregate, label, and put the product on quarantine—and someone must confirm that all of the affected material has been put on hold.
Informing Proper Parties
Any serious issues concerning distributed product must be reported to FDA quickly. A company may also need to perform a recall or a correction and removal. If a recall becomes necessary, consult with FDA to determine the recall classification and to discuss the strategy for conducting it. Although industry and FDA sometimes disagree on what constitutes a reportable event, it is better to err on the side of caution and report problems to the agency.
Information concerning CAPA, nonconforming products, and quality problems should also be disseminated internally, including to senior management. A clearly defined risk management process should be in place for elevating key items to senior management. Some companies may categorize risk by levels or by terminology (such as a critical, major, or minor risk—a critical item being one that can lead to possible patient injury or FDA regulatory action). Many firms place a QA executive or management representative in charge of immediately notifying senior management about serious quality issues and ongoing investigations. Routine information about quality system performance can be reported to management in periodic management reviews, monthly reports, weekly group meetings, etc.
It is a regulatory requirement to inform senior management about quality issues, the results of investigations, and FDA inspectional observations. Senior management should be aware of quality problems and should help a firm take prompt, appropriate action. There are a number of ways management can help. Management should ensure that sufficient funding is available and that sufficient resources are available, make it a priority to resolve the matter, and set an example by always doing the right thing.
Conclusion
Depending on the maturity of a CAPA system, it may take some time to get it into the shape it should be. Bear in mind that a good way to start is by improving any critical deficiencies first. Many companies in the medical device industry will honestly admit that they are not yet pleased with (and are continually improving) their CAPA system. A company's CAPA system gets to the heart of all its employees' ability to think critically, to the quality and caliber of its QA executive and staff, and to the organization's commitment to the quality of its products.
References
1. Federal Register, 61 FR:52601, October 7, 1996 [online]; available from Internet: www.fda.gov/cdrh/fr1007ap.pdf.
2. ANSI/ISO/ASQ Q9000-2000, “Quality Management Systems—Fundamentals and Vocabulary” (Milwaukee: American Society for Quality, 2000).
3. “Points to Consider When Preparing for an FDA Inspection under the QSIT Corrective and Preventive Action Subsystem” [online] (Washington, DC: AdvaMed, 2001); available from Internet: www.advamed.org/publicdocs/pointstoconsider6-5-01.pdf.
4. GHTF/SG3/N15R8, “Implementation of Risk Management Principles and Activities within a Quality Management System” [online] (Global Harmonization Task Force, 2005); available from Internet: www.ghtf.org/sg3/inventorysg3/sg3n15r82005.pdf.
5. Dean L Gano, Vicki E Lee, and Wendy C Mitchell, Apollo Root Cause Analysis—A New Way of Thinking (Mansfield, OH: Apollonian Publications, 1999).
6. “FDA Guide to Inspection of Quality Systems” [online] (Rockville, MD: FDA, 1999); available from Internet: www.fda.gov/ora/inspect_ref/igs/qsit/qsitguide.pdf.
7. Linda Bren, “Investigators' Reports: Company Caught in Coverup of Medical Device Malfunctions,” [online] FDA Consumer magazine 37, no. 6 (2003); available from Internet: www.fda.gov/fdac/departs/2003/603_irs.html.
8. FDA warning letter to James M. Cornelius, Guidant Corp. [online] 22 December 2005; available from Internet: www.fda.gov/foi/warning_letters/g5657d.pdf.
Barbara K. Immel is president of Immel Resources LLC (Petaluma, CA) and can be e-mailed at [email protected].
Copyright ©2006 Medical Device & Diagnostic Industry
About the Author(s)
You May Also Like
.png?width=300&auto=webp&quality=80&disable=upscale)
.svg?width=300&auto=webp&quality=80&disable=upscale)
