Sign up for the QMED & MD+DI Daily newsletter.
What's Behind Medtech's Recall Epidemic? Part 3: Production Controls
Problems related to production controls account for almost a quarter of medical device recalls, and three failure modes are to blame in more than two-thirds of cases.
July 18, 2016
6 Min Read

Problems related to production controls account for almost a quarter of medical device recalls, and three failure modes are to blame in more than two-thirds of cases.
Joshua R. Dix, Suraj Ramachandran, and Darin S. Oppenheimer
Editor's Note: This is the third installment in a four-part series on the factors behind medtech's recall epidemic. Read Part 1 and Part 2.
When you take into consideration that elements of the manufacture and servicing of medical devices make up 24% of the controls set in place by FDA's 21 CFR 820 Quality System Requirements, it should come as no surprise that recalls related to these same controls make up 24% of all recalls since 2010. As we have done with both design and software controls before it, our analysis of production controls seeks to explore the specific recalls, hazardous situations, trends, and possible mitigations related to each of the root causes associated with production control related recalls.
While the controls set forth by FDA in the 1978 Food, Drug, and Cosmetic Act (amended in 1990 to include design controls) aimed at providing consumers with safer and more effective products, recalls related to production controls have accounted for an average of 665 recalls per year since 2010, with an annual growth rate of 11% percent over the same five year span, according to Blue Lynx Consulting (Figure 1).
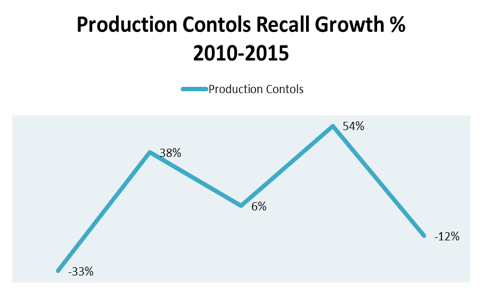
Figure 1. Production controls growth perent by year, 2010-2015.
Like the analysis of both design and software controls, the upward trends related to production controls point to a reactive industry culture that continues to face issues with adverse events despite legislative oversight and guidance.
Recalls Related to Production Controls
In comparison with the astronomical growth rates of the other two leading causes of medical device recalls (device design (+334%) and software controls (+225%), those related to production controls are growing at a much slower rate. While some members of industry may view this as a positive, production controls continue to be the second leading cause of medical device recalls on a yearly basis (Figure 2).
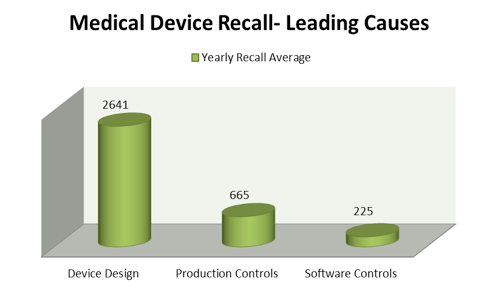
Figure 2. Leading device recall causes 2010-2015.
Furthermore, the year-to-year fluctuations (Figure 3) of both production control recalls and their subcategories demonstrates the inability of industry to adequately control these recalls or set into place the proper mitigations to prevent their growth.

Figure 3. Produciton control recall subcategories growth rate percent 2010-2014.
Specifically, the giant swings in areas of production controls like errors in labeling (Figure 4) and packaging (Figure 5) exhibit industry's inability to control adverse events in portions of business which should be easy wins.
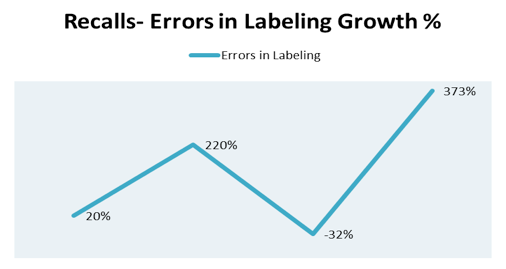
Figure 4. Recalls-Errors in labeling rowth percent by year 2010-2014.
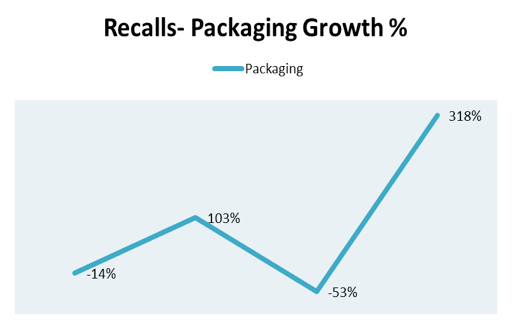
Figure 5. Recalls-Packaging growth percent by year 2010-2014.
Why Do Recalls Related to Production Controls Reoccur?
While the fluctuations in yearly growth percentages related to production control recalls may be vast, three failure modes consistently account from more than 65% of all manufacturers' recalls of the same category. Specifically, recalls related to process controls, packaging, and packaging controls made up an average of 68% of all production control recalls from 2010 to 2015, with bookend highs coming in 2010 (77%) and 2015 (76%).
As a result, any hope the medical device industry has at stemming future issues related to production controls must begin with a tactical plan aimed at reducing the hazards associated with these specific failure modes and working outwards throughout the year without losing focus on other known issues which are potentially harmful to both the business and end user.
In the inaugural article of this series, we asked why issues reoccur. Throughout the subsequent analysis, the statistical evidence demonstrated that these issues do not only reoccur; the existence of the reoccurrence points to a more severe and altogether disconcerting problem. When more granular analysis is applied to the reoccurrence of these adverse events, evidence unveils industry's inability to demonstrate year-to-year control over their products.
Sadly, the history of the medical device industry parallels Newton's Third Law of Physics. For every Cooper Committee, there is a Therac 25 tragedy; for every 21 CFR 820, there is a Super Bug outbreak. The question remains: Does it have to be this way? As we close the chapter on the hazardous situations and trends related to the United States' recall epidemic, we will revisit the question of why issues reoccur and provide further insight into the possible mitigations that could lead to the end of these problems.
Disclaimer: The perspectives shared in this article represent the personal opinions and insights of the individual authors and are not associated with the authors' respective employers.
Note: All supporting statistical information garnered from the FDA Recall Database in support of this article was compiled and provided by Blue Lynx Consulting.
Joshua R. Dix is a regulatory affairs specialist. Based in the Western New York area, he is the global regulatory lead for multiple product platforms with responsibilities including regulatory strategy, submissions, product CAPA, and audits. With extensive experience in both regulatory affairs and quality systems, Dix has worked to bring multiple medical devices to market in more than 20 different countries. He holds a bachelor's degree in English from the State University of New York.
Suraj Ramachandran, MS, is a regulatory affairs manager. Based in the Chicago area, he is involved primarily with managing the infusion pump platform and supporting all new product development and life cycle maintenance activities, including regulatory submissions, design control, audits, and CAPAs. Ramachandran has also led many development efforts regarding medical device software intended for both domestic and international markets. He holds an undergraduate degree in biomedical engineering as well as a master's degree in biomedical engineering from the University of Michigan. In addition, he holds a RAC certification from the Regulatory Affairs Professional Society.
Darin Oppenheimer, MS, is a director of regulatory affairs. He is involved in many facets of the product development life cycle, including regulatory submissions, due diligence, and active participation on industry trade organizations and standards committees. Oppenheimer leads a team of regulatory professionals focusing on electromechanical devices and software. His prior background as a research and development scientist focused on pharmaceuticals and medical device diagnostic applications for biomarker and drug discovery. His undergraduate degree is in molecular biology from the University of Tampa. He also holds two master's degrees from Johns Hopkins University in biotechnology and regulatory science, as well as a graduate certificate in biotechnology enterprise.
[image courtesy of NIYAZZ/iSTOCKPHOTO.COM]
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
