Sign up for the QMED & MD+DI Daily newsletter.
Regulatory Guidelines For Biocompatibility Safety Testing
Medical Plastics and Biomaterials Magazine MPB Article Index Originally published May1997 BIOCOMPATIBILITY
15 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Regulatory Guidelines For Biocompatibility Safety Testing")
BIOCOMPATIBILITY
Biomaterials and medical devices constitute an extremely diverse, heterogeneous category of items. Because the use of these products normally entails their direct or indirect contact with patients, there is an obligation on the part of manufacturers to establish the safety of their products before they are marketed. Medical device safety evaluation assesses the risk of adverse health effects due to normal use and likely misuse of a device. Since adverse health effects could result from exposure to the materials from which a device is made, preclinical assessment of the toxic potential of such materials or components is needed to minimize the potential hazard to the patient.
 A thorough biocompatibility safety testing program will typically comprise in vivo studies supplemented by select in vitro assays. Photo: Northview Biosciences, Inc.
A thorough biocompatibility safety testing program will typically comprise in vivo studies supplemented by select in vitro assays. Photo: Northview Biosciences, Inc.
Until recently, the regulations governing the manufacture and sale of medical devices varied greatly among countries. Since January 1995, medical devices to be marketed in the European Union (EU) have been required to comply with EU Medical Devices Directive 93/42/EEC, which specifies requirements for safety assessment issues. The purpose of the directive is to promote a single European market for trade in medical devices, while ensuring that users and patients are not exposed to unnecessary risks.
At present, safety assessments of medical devices are guided by the toxicological and other studies recommended in the International Organization for Standardization (ISO) 10993-1/EN 30993-1 standard. At present, 17 parts of the standard are either accepted or under preparation. Tests that may be used in an evaluation of medical device biocompatibility include procedures for cytotoxicity, skin sensitization, dermal irritation and intracutaneous reactivity, acute systemic toxicity, subchronic toxicity, mutagenicity, implantation, hemocompatibility, chronic toxicity, and carcinogenicity.
This article is an introduction to a relatively new and rather complicated field in toxicology—the toxicological testing of medical devices. The guidelines for testing such products are discussed and a general description of the various test procedures given. Future developments in the field of biocompatibility regarding international harmonization and the potential for new methodologies are also addressed.
INTERNATIONAL REGULATORY EFFORTS
ISO is in the process of publishing a series of standards on the biological evaluation of medical devices—ISO 10993.1 Many parts of this series have been accepted as international standards, while the rest are under development (see Table I). The subject of the first part, ISO 10993-1, is the categorizing and performance of safety testing. Part two of the standard, ISO 10993-2, is concerned with animal welfare requirements; another section, ISO 10993-12, deals with sample preparation and reference materials. Most of the remaining parts of the standard treat the individual tests.
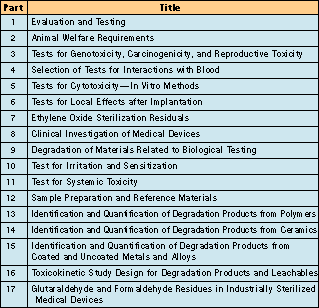 Table I. Listing of individual parts of ISO 10993, Biological Evaluation of Medical Devices.
Table I. Listing of individual parts of ISO 10993, Biological Evaluation of Medical Devices.
The EU has issued a council directive—93/42/EEC, 1993—concerning medical devices.2 All medical devices to be sold on the EU market must comply with this directive after June 14, 1998. The European Committee for Standardization (CEN) is currently in the process of adopting the ISO 10993 standard as the European standard.
In 1986 the responsible authorities in the United Kingdom, United States, and Canada issued the Tripartite document, which was a guidance on the selection of toxicological tests for medical device safety testing.3 This document has now been replaced by ISO 10993-1 as a first step in the process of international harmonization. In 1995 FDA chose to accept the ISO 10993-1 standard, with a modification of the matrix listing (see sidebar below).
Japanese authorities have also issued a guideline for toxicological testing of medical devices. This document is available in an unofficial translation as Guidelines for Basic Biological Tests of Medical Materials and Devices.4 It resembles ISO 10993 in structure and content, but recommends modified tests and sample preparations.
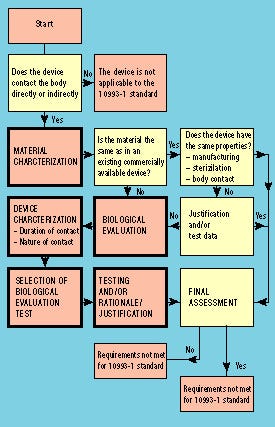 Figure 1. Flowchart illustrating steps in the biological evaluation of medical devices according to the ISO 10993-1 standard.
Figure 1. Flowchart illustrating steps in the biological evaluation of medical devices according to the ISO 10993-1 standard.
The procedure for using the ISO 10993-1 standard is illustrated by the flowchart in Figure 1. The standard is applicable only for devices that are directly or indirectly in contact with the body or body fluids. If a device is to be subjected to the standard, the first step is to characterize the material. Such characterization need not always be followed by biological evaluation, because there may be sufficient historical data to verify that the device meets the requirements of the standard. If the material and/or the intended use of the device is different from any historical safe device, biological evaluation has to be performed. By following the standard, a suitable test program can be chosen depending on the type and duration of body contact.
Within the EU, all new medical devices must carry the CE mark from June 14, 1998. This should ensure the availability of relevant documentation regarding biocompatibility and the lack of health problems associated with the use of a device. It is noteworthy that the approval of such documentation is not, as it was previously, accorded by the national health authorities, but rather by the so-called notified bodies, whose experts review the products and production facilities of medical device manufacturers.
DEVICE CATEGORY AND CHOICE OF TEST PROGRAM
The need to evaluate a medical device biologically depends on the material used in the device, the intended body contact, and the duration of that contact. A device designed for surface contact for a limited time is not as likely to be bioincompatible as a permanent-exposure implant device made of the same material. The ISO 10993-1 standard divides medical devices into three main categories: surface devices, externally communicating devices, and implant devices. Each category is further divided into subcategories according to the type of contact to which the patient is exposed (see Table II).
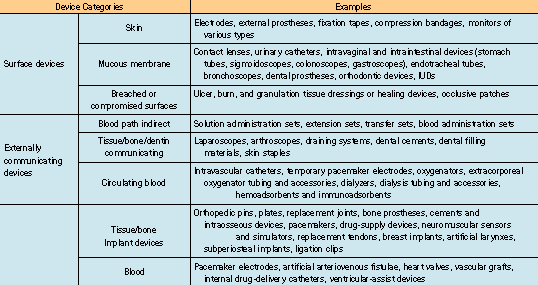
Table II. Device categories and examples according to ISO 10993-1.
The choice of test program for a device in a given category depends on the duration of the contact. Three different time spans are given: limited contact (<24 hours), prolonged contact (24 hours30 days), and permanent contact (>30 days). ISO 10993-1 lists the tests that must be considered for each category (note: the accompanying table could not be reproduced. Please contact the editors for more info).
The ISO test matrix should not be considered as a checklist for the different tests that have to be performed, but rather as a guide for qualified toxicologists who also take into consideration material information and historical data from similar devices. The certifying authorities in most countries (e.g., notified bodies, FDA, Japanese authorities) are generally cooperative when a company must decide on a test program for a device. It is therefore advisable to maintain close contact with the relevant authorities during the entire process. However, testing should not be performed simply to meet regulatory requirements. This is important not only to lessen the risk of overtesting and excessive use of experimental animals, but also because a strict regulatory approach may mask potential negative health effects that might be identified via optional or nonroutine testing procedures.
As regards CE marking of existing products on the market or safety evaluation of medical devices already in clinical use, appropriate historical or clinical data should be employed whenever possible to avoid unnecessary testing.
PREPARATION OF EXTRACTS
ISO 10993-12 describes how samples for biological evaluation should be selected, prepared, and extracted. Other guidelines provide similar descriptions, which differ slightly in the specifics of the extraction procedures.
The device to be tested (the test article) should be a representative specimen of the mass-produced device. It should also be finished or treated (e.g., coated or sterilized) in the same way as the mass-produced device.
Because the toxic potential of materials and devices depends to a substantial degree on the leachability and toxicity of soluble components, extracts of the device are normally used in the tests. In some tests, however, an evaluation under normal-use conditions is mimicked by using the device or a piece of the device directly. Ideally, extraction media should constitute a series of media with decreasing polarity to ensure the extraction of components of widely different solubility properties. The most commonly used extraction media are physiological saline, vegetable oil, dimethylsulfoxide, and ethanol. Other extraction media such as polyethylene glycol or aqueous dilutions of ethanol may be selected in certain cases. For in vitro cytotoxicity testing, complete cell-culture medium is most often employed.
The various guidelines also differ somewhat with respect to the temperature at which the extraction is conducted. Some leachable compounds may be chemically altered at high temperatures, and it is now generally recommended that extraction be conducted at 37°C—simulating body temperature—for 72 hours. This procedure will probably become increasingly accepted as the most appropriate extraction method. For in vitro cytotoxicity tests, extraction at 37°C for 24 hours is usually recommended, since certain constituents of the media are relatively labile.
The amount of leachable substances released to the extraction media is related to the surface area and thickness of the product to be extracted. Recommendations vary from 1.25 to 6 cm2 of product per milliliter of extraction medium, depending on the size and shape of the product, or from 0.1 to 0.2g of product per milliliter of extraction medium when a surface area cannot readily be estimated (e.g., for powders or granulates). In any case, the specific properties of the product must be taken into account in order to make usable extracts.
For cases in which a medical device comprises several components made from different materials, the ideal procedure from a toxicological point of view would be to test extracts of the components separately. However, in some situations this is not practical, and extracts of the whole device may be used instead.
BIOLOGICAL CONTROL TESTS
Biological control tests are not described in the ISO 10993 standard for biological evaluation of medical devices, since these particular tests are designed primarily for batch-control purposes. Such tests are also used during the product development phase to identify sources of contamination and to establish procedures that ensure the intended quality of the end product.
Microbiological Control Tests. Microbiological control tests are necessary to establish the microbiological status of an end product—factors such as sterility, absence of pathological bacteria, or limits for microbial counts. Furthermore, it is often necessary to monitor the microbiological load of raw materials and intermediary products, or to check the efficiency of production and sterilization processes. The tests are performed by rinsing the materials or products in physiological saline and assessing the rinsing medium for microbes, or by directly incubating the products in growth media. Although the presence of pathological microbes on medical devices can represent a potential health problem, the subject is not within the scope of this article.
Tests for Endotoxins. Even sterile medical devices may contain cell-wall lipopolysaccharides originating from gram-negative bacteria. Such so-called endotoxins or pyrogens can cause an abrupt fever reaction after entering directly into the body from sources such as venous catheters, syringes, or implant components.
Two different biological assays can be used to measure the presence of endotoxins: the rabbit pyrogen test and the Limulus test. In both cases, an eluate is prepared—normally by rinsing the surfaces of the product with water—and then tested for endotoxins. In the rabbit pyrogen test, the eluate is injected intravenously and the rectal temperature of the animal is measured after the injection. In the Limulus test, the eluate is incubated together with lysate from the blood of the horseshoe crab (Limulus polyphemus), which contains a substance that forms a gel in the presence of endotoxins.
Test for Nonspecific Toxicity. This test is designed to assess any nonspecific adverse effect that occurs following intravenous injection of a device eluate in mice. The test is often performed with the same eluate used for the pyrogen test. The mice are inspected regularly for any signs of ill health, which can indicate the presence of toxic substances leaching from the product.
In May 1995, FDA issued Blue Book Memorandum G95-1, Use of International Standard ISO-10993, "Biological Evaluation of Medical Devices Part 1: Evaluation and Testing." This memo was a huge step toward international harmonization of device biocompatibility testing, although there are still some significant differences between FDA and European requirements. The Blue Book supersedes the Tripartite Guidance as a guideline for planning biocompatibility testing of medical devices for the U.S. market. The most significant change from the Tripartite is FDA's increased emphasis on a case-by-case testing strategy for individual devices. The biocompatibility of a device depends on several factors, especially the type of patient tissue that will be exposed to device materials and the duration of the exposure. Neither the Blue Book memo nor ISO 10993 prescribes a specific battery of tests for any particular medical device. Rather, they provide a framework that can be used to design a biocompatibility testing program. As with the Tripartite and the ISO standard, the core of the Blue Book memo is a materials biocompatibility matrix. The matrix categorizes devices based on the type and duration of tissue contact. It also presents a list of potential biological effects. For each device category, certain effects must be considered. In preparing a regulatory submission, manufacturers must address each of the biological effects pertinent to their device. The matrix from the Blue Book memo incorporates several ISO features:
The test-selection matrix applies to most premarket approval submissions. The Blue Book memo also contains a flowchart to assist in selecting toxicity tests for 510(k) devices. If there is no significant change between the new device and the predicate device in materials, manufacturing, sterilization methods, and body contact, no further biocompatibility testing may be necessary. Otherwise, manufacturers are referred to "Device Specific Tox Profiles" (to be issued by FDA at some yet-to-be-determined date) or to the Blue Book matrix. When designing a biocompatibility testing program, manufacturers should also consult other applicable FDA publications, such as Biocompatibility of Medical Devices (from the Center for Devices and Radiological Health), the Guidelines for the Intraarticular Prosthetic Knee Ligament, or the PTCA Catheter System Testing Guideline. Other useful documents include ASTM F 748, Practice for Selecting Generic Biological Test Methods for Materials and Devices, and the AAMI Standards and Recommended Practices, Volume 4: Biological Evaluation of Medical Devices. (These and other publications are available from CDRH Facts-On-Demand at 800/899-0381.) For most projects, companies should review testing plans with FDA before beginning the actual studies. Since the Blue Book memo was issued, a few trends have emerged:
So what's the bottom line for manufacturers planning biocompatibility testing programs? Work with an experienced toxicologist to apply the Blue Book matrix and the ISO standard to your device. Avoid cookie-cutter solutions by carefully evaluating the intended use of the device. And if at all possible, consult your FDA reviewer before beginning your biocompatibility testing program. RELATION TO U.S. PHARMACOPEIA To test medical device biocompatibility, manufacturers often use USP procedures such as the USP in vivo biological reactivity tests (Class I–VI plastics tests). While class plastics tests have some value in a biocompatibility testing program, a full Class VI test is rarely needed for a medical device. As a general rule, the Blue Book memo and ISO documents take a broader and more thorough view of biocompatibility than does the U.S. Pharmacopeia, and they supersede the USP for evaluating which studies to submit to FDA in support of product registrations. SOURCES OF BIOCOMPATIBILITY TEST DATA Data to satisfy biocompatibility test requirements may come from any of several sources. Most commonly, companies arrange for their own biocompatibility studies. Material vendors are often willing to share test data they have generated. If vendor data are used, manufacturers should obtain copies of the original study reports, and they should conduct at least some confirmatory testing of their own (e.g., cytotoxicity and hemocompatibility studies). If available, clinical data can be used to satisfy some biocompatibility biological-effect categories in the matrix. Manufacturers may use analytical data (e.g., extraction studies) to eliminate the need for biological testing in a particular category. Note on Contributors: Timothy V. Doherty, DVM, is director of in vivo services and Jeffrey Wallace is manager of toxicology services at the Northview Pacific Laboratories (Berkeley, CA) division of Northview Biosciences, Inc. (Northbrook, IL). The information is derived from the company's publication entitled Assessing Biocompatibility: A Guide for Medical Device Manufacturers. |
About the Author(s)
You May Also Like


