Sign up for the QMED & MD+DI Daily newsletter.
Postmarket Surveillance: The Truth Is Never Simple (Part 2—Manufacturer Considerations)
In the second installment of this three-part article series, experts discuss federal initiatives regarding medical device postmarket surveillance and set out what device manufacturers should know.
December 16, 2016
6 Min Read
")
In the second installment of this three-part article series, experts discuss federal initiatives regarding medical device postmarket surveillance and set out what device manufacturers should know.
Kevin Ong, PhD, PE, Michael Frohbergh, PhD, Jennifer Stevenson, Esq., and John Constance, Esq.

Editor's note: This is the second installment in a three-part series detailing the ins and outs of medical device postmarket surveillance. Read Part 1 and Part 3.
Signal Detection
While FDA expects manufacturers to perform trend analysis as part of their complaint handling system, manufacturers may also perform additional signal analyses on information collected from other postmarket surveillance sources. Signals can include unexpected safety hazards and failure modes, high severity events, nonconformance and malfunctions, statistical process control, and trend signals. "High severity" events may depend on the product; for example, hospitalization following the use of a low-risk product, such as a hearing aid, may raise more concerns than following the use of a high-risk product, such as a cardiovascular device for treating a life threatening condition.
Statistics are often used to determine the presence of a signal in terms of outliers or "blips" in product performance. Statistical process control utilizes historical data to first determine what is considered to be statistically "likely." Based on the variability (i.e., standard error) in the historical data, the threshold ("control limits") at which the performance is considered statistically "unlikely" is identified. If the frequency of adverse events in a given period exceeds the control limit, this indicates the presence of a signal. Even in the absence of a signal, upward trends in complaint frequency may raise concern and trigger more scrutiny. Ultimately, as appropriately stated by FDA, "[d]ata mining analyses are used to detect potential signals and generate related hypotheses, but cannot be used in isolation to establish causality."
Ongoing Federal Initiatives
FDA's vision for postmarket surveillance is an integrated health system that will enable timely, accurate, systematic, and prioritized assessments of the benefits and risks of medical devices. It is expected that this system will include a wide variety of data sources, including 522 postmarket surveillance studies, post-approval studies, registries, and MDRs, and that it will eventually fully incorporate unique device identifiers (UDIs) (described below). By using this system, FDA aims to identify potential safety signals in near real-time to determine the performance of devices already on the market, as well as to facilitate the clearance and approval of new devices or new uses for existing devices.
Unlike drugs, medical devices cannot always be easily identified at the level of specificity necessary for effective safety surveillance. Seeking to remedy this limitation, in 2007, Congress passed legislation requiring that FDA develop regulations to establish UDIs in order to more accurately track, monitor, and assess the performance of medical devices. UDIs are codes assigned to devices that identify the specific version or model of a device, its lot/batch numbers, and production and expiration date information. FDA intends that UDIs will be standardized identifiers for linking electronic health records, registries, and hospital claims data. They will allow for the facilitation of adverse event reporting, the ability to more effectively manage recalls, and identification of counterfeit products. Compliance with this legislation started as early as September 2014 for Class III devices with a completion goal of late 2020 for lower-risk devices.
In addition, FDA introduced the Sentinel Initiative, a national integrated, electronic system to monitor medical product safety. The Initiative aims to provide active product surveillance by leveraging existing healthcare information from electronic health records, insurance claims data, and patient registries. Initial efforts have been made through the Mini-Sentinel pilot project, which launched in 2009 and, as of August 2015, utilizes 18 data partners with access to data for 193 million individuals. FDA is awaiting full UDI implementation to provide more useful medical device data for the Sentinel Initiative.
Considerations for the Manufacturer
Manufacturers, importers, and user facilities should be cognizant of regulatory requirements for postmarket surveillance and implement effective strategies to comply with these regulations, while also maintaining their research and development efforts. Adopting a proactive approach to performance and process monitoring can ultimately save the manufacturer significant time and expense. Potential monitoring may include detecting manufacturing issues, identifying changes in performance trends, evaluating customer satisfaction, periodic reviewing of risk analyses for unanticipated misuse or failure modes, and collecting periodic feedback on the instructions for use from users and any training needs.
Before undertaking postmarket surveillance efforts, manufacturers should carefully consider what data sources, metrics/benchmarks for detecting signals, and postmarket review cycles are appropriate. The potential data will vary based on the type of product, the specificity of product identification in the data, and the quality of the data, to name a few. Depending on the data source, the frequency of data collection and review cycles maybe ongoing, monthly, quarterly, etc.
Additionally, data collection and review may not necessarily occur at the same frequency. For example, product complaints and returned products may be collected on an ongoing basis as they are received, but reviews may occur on a monthly basis. The scrutiny and frequency of manufacturer monitoring depends heavily on device type, risk classification, novelty of the technology, market experience, and the availability of scientific knowledge.
Moreover, consideration should be given to the metrics or triggers (control limits/outliers, trend over a particular period of time, etc.) that are used to indicate a signal. Determining the appropriate benchmark for an outcome can be challenging due to variations in patient populations and end use conditions. Should outcomes be compared between generations of a device? If so, how should one account for possible expansion of the use indications between the device generations in the analysis? Should outcomes also be considered between similar products made by their competitors?
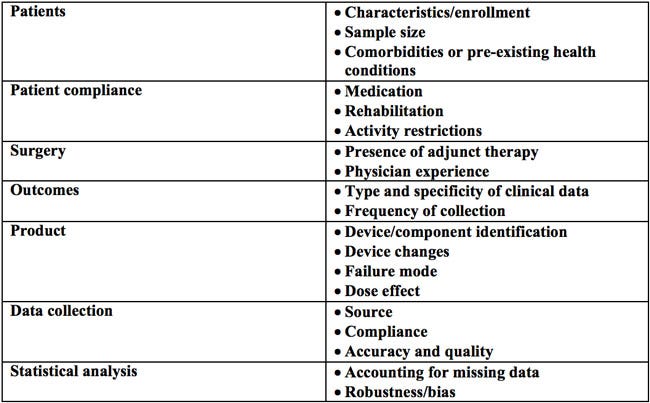
The type of analytical approach or data mining methods can vary from more basic to highly sophisticated techniques. Finding the right balance in the complexity of the approach can be difficult. Basic approaches may not provide adequate filtering of noise versus true signals. On the other hand, more complex methods may lead to the equivalent of paralysis by analysis due to over-analyzing the data or over-complicating the decision-making process. Interpreting information from these various sources requires critical consideration of a number of variables (Table 2).The recommended approach in any given situation will likely be guided by general industry practices and the type of product involved.
Table 2. Factors to consider when interpreting source information
Kevin Ong, PhD, PE, is a principal engineer in the biomedical engineering practice at Exponent, Inc, an engineering and scientific consulting firm.
Michael Frohbergh, PhD is an associate in the biomedical engineering practice at Exponent, Inc.
Jennifer Stevenson, Esq. is a partner at Shook, Hardy & Bacon, LLP in Kansas City.
John Constance, Esq. is an associate at Shook, Hardy & Bacon, LLP in Washington, DC.
[Image courtesy of CLKER-FREE-VECTOR-IMAGES/PIXABAY]
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
