.jpg?width=700&auto=webp&quality=80&disable=upscale "Ortiz_table_1(1).jpg")
Sign up for the QMED & MD+DI Daily newsletter.
FDA Focus: Managing Supplier Purchasing Controls
FDA recently told manufacturers that they bear responsibility for every step of their global supply chain. Recent recalls of medical devices due to failures of critical components and services supplied to device manufacturers prompted FDA’s increased scrutiny of supplier purchasing controls.
+2
20 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "FDA Focus: Managing Supplier Purchasing Controls")
FDA recently told manufacturers that they bear responsibility for every step of their global supply chain. Recent recalls of medical devices due to failures of critical components and services supplied to device manufacturers prompted FDA’s increased scrutiny of supplier purchasing controls.
How serious is the agency? In 2009, 12% of 483 observations and 16 warning letters issued were all related to inadequate supplier qualification.1
Additionally, Megan Moynahan, network leader for the Cardiac Electrophysiology and Monitoring Devices Network at CDRH, stated that with the support of the consulting firm McKinsey & Company, CDRH developed a signal and escalation program designed to connect the dots for end-product or component quality issues within and between industry segments. This program will improve the agency’s ability to quickly investigate and take action to protect public safety. For example, if a battery component quality issue were discovered in the inspection of one pacemaker, the agency would look at its potential effect on other products for which the supplier provides batteries, in addition to its potential effect on all pacemakers.
FDA is not only focusing on U.S. manufacturers and suppliers, but also overseas suppliers. With more than 700 recently hired inspectors, the agency plans to ensure that it performs overseas inspections on a timely basis.2
What does this mean for OEMs? Now more than ever, OEMs need to perform a formidable balancing act between risk, cost, and quality, while maintaining regulatory compliance. Moving forward, OEMs need to think of their suppliers as if suppliers were part of the their in-house production facility. This article provides a clearer understanding of how industry reached this point. It outlines some trends and compliance expectations and offers practical solutions for manufacturers and suppliers.
How We Got Here
Critical components, like printed circuit boards, were once manufactured internally by OEMs. An increase in failures of these components led to recalls and patient safety issues and caught the attention of FDA and Congress.
Prior to implementing the medical devices current good manufacturing practice (cGMP) final rule quality system regulation (21 CFR Part 820) in 1996, FDA solicited and addressed industry comments on the proposed regulations. Much of the feedback focused on purchasing and supplier controls. Industry expressed concerns that application of the regulation to component manufacturers would increase product cost, with questionable value added to device safety and effectiveness. It was feared that many component suppliers would refuse to supply components or services to the medical device industry, especially those for whom the industry accounted for just a small fraction of sales.
FDA stated that because of the complexity of components used in medical devices, each component’s adequacy could not always be assured through inspection and testing at the OEM. Instead, the quality of a component or service needed to be established during the design of that component or service, and achieved through implementing proper control and quality systems as part of the supplier’s manufacturing process.
The proposed quality system regulation (QSR) explicitly required that the finished device manufacturer assess the capability of suppliers, contractors, and consultants to provide quality products pursuant to Sec. 820.50 purchasing controls. These requirements supplement the acceptance requirements under Sec. 820.80. As a result, FDA agreed with industry and removed the provision making the cGMP regulation applicable to component manufacturers.3
However, FDA noted that it would continue to focus its inspections on finished device manufacturers and would expect that OEMs properly ensure that the components they purchase are safe and effective. Finished device manufacturers that fail to comply with Secs. 820.50 and 820.80 would be subject to enforcement action. FDA also noted that the legal authority exists to cover component manufacturers under the cGMP regulation, should the need arise.3
As a result, the manufacturer must determine the adequacy and extent of required quality systems, with particular emphasis in change control and corrective and preventive action (CAPA), through auditing and other verification or assessment tools. FDA indicated that the degree of supplier control necessary to establish compliance may vary with the type and significance of the product or service purchased and the effect of that product or service on the quality of the finished device.3
Sec. 820.80 is specific to a device manufacturer’s acceptance program. Although finished device manufacturers are required to assess the capability of suppliers, contractors, and consultants to provide quality products and services, inspections, and tests, other verification tools are also an important part of ensuring that components and finished devices conform to approved specifications. The extent of incoming acceptance activities can partially be based on the degree to which the supplier has demonstrated a capability of providing quality products or services. 3
An appropriate component supplier and services quality assurance program includes a combination of assessment techniques, including inspections and tests. Manufacturers should remember that the purpose of assessing a supplier’s capability is to provide a greater degree of assurance beyond what provided by an inspection and test. The process ensures that the products received meet the finished device manufacturer’s requirements.3
With regard to change control, industry was concerned with the requirement in Sec. 820.50(b) mandating that suppliers notify OEMs of any change in their component or service. OEMs were concerned that this placed an undue burden on suppliers and inhibited their ability to make minor adjustments within the parameters of agreed upon specifications and quality requirements. Additionally, many people in the industry believed that the requirement was feasible only for components that were custom-made for the OEM, and was meaningless with regard to commercial off-the-shelf components purchased from distributors. Others were concerned that suppliers would not be willing to supply device manufacturers with such information. However, others commented that the requirement was too broad and would result in burdensome reporting of variables that were irrelevant to the continued performance or specifications of the component or service. FDA agreed in part with the comments and amended the requirement to state that such agreement should be obtained where possible. Even after making this accommodation, FDA stated that it still believed that it was important for the manufacturer to obtain information on changes made to the component. A supplier that refuses to provide such notification may be rendered unacceptable, depending on what they supply. However, when the product is in limited supply and must be purchased under those conditions, the manufacturer must heighten control in other ways (e.g., tightening incoming inspection or downstream controls prior to distribution). FDA gave manufacturers the flexibility to define in the agreement the types of changes that would require notification.3
Outsourcing to Suppliers
Establishing robust and clear supplier agreements continues to be a challenge for industry. Even with these agreements in place, many changes that seem innocuous when first implemented may later manifest as significant issues in distributed products. This is caused in part by weak supplier agreements that do not properly define notification requirements or apply sufficient oversight from the OEM.
Concerned with ongoing supplier controls issues,as well as a desire to harmonize regulations and standards across the world, Kim Trautman, FDA’s medical device expert and coauthor of the quality systems regulations, participated in developing the Global Harmonization Task Force’s (GHTF) guidance on the subject, titled, “Quality Management System—Medical Devices—Guidance on the Control of Products and Services Obtained from Suppliers.” The guidance was published in late 2008.4 Although Trautman said that the GHTF document cannot be enforced as a regulatory compliance requirement, it is clear from her presentations to industry that the agency views the guidance document to be aligned with the QSR (see Table I).
|
Table I. Click for enlarged view. |
Regulatory Enforcement Trend
One recent enforcement trend is supplier process validation. As the GHTF guidance document indicates, “Regulatory requirements call for processes to be validated where the resulting output cannot be verified by subsequent monitoring or measurement. Regardless of who actually performs the process validation, it is the manufacturer’s responsibility to ensure that the validation is properly performed, and that it cross-references the GHTF Guidance on Process Validation SG3/N99–10:2004.
There is another important consideration to be made regarding validation requirements performed on supplier processes. It is imperative that OEMs ensure that validation processes employed by their suppliers meet the minimum requirements of the validation processes of the OEM as part of either the qualification of the supplier by the OEM, or through the review of validation protocols conducted by the supplier for the manufacturer. This process would include all aspects of validation, including process output performance levels (i.e., acceptance criteria), statistical requirements, review or resolution of deviations, etc.
To ensure that the validated process continues to operate within a state of control, manufacturers must work with suppliers to establish critical controls, limits, and an ongoing review of process control data. Remember, the supplier may own the process, but the manufacturer owns the product. Major quality systems areas include
? Quality management.
? Personnel.
? Buildings and facilities.
? Documentation and records.
? Purchasing controls.
? Production and in-process controls.
? Validation.
? Change control.
? Complaints and recalls.
Drawing the Applicability Line
The criticality of the component from a product-function and patient-safety risk perspective plays an important role. However, a noncritical component could be contaminated through supplier processing and shipping, which could lead to patient injury or death.
Based on enforcement trends, the base applicable quality systems that all suppliers should have are change control (design and process); process control (including process validation where the product quality attributes including stability cannot be fully verified) and supplier quality assurance for their critical raw material suppliers. Other aspects of a OEM’s quality system, such as complaint handling or statistical requirements, may also apply.
For commodity components that are commercial and off the shelf, the OEM should verify that the supplier puts in place the relevant industry test standards that apply to the component. If a supplier is unwilling to comply with the base quality systems described, the OEM should look for a new supplier that will comply. The OEM should also implement tightened incoming quality assurance inspection or downstream verification during production of the finished product as an interim or permanent control while searching for a new supplier.
Ultimately, manufacturers need to think of their suppliers as part of their in-house production facility. When applicable, the supplier’s procedures must meet all of the regulatory and specific product requirements of the manufacturer’s quality system.
Theoretic vs. Pragmatic Realities
These procedures apply to all suppliers and all components, including other divisions within the same company. In industry forums, FDA has indicated that it is unacceptable to use the corporate audit of a sister division to verify that the supplier has adequate quality systems. These audits are often focused solely on compliance with corporate standards and policies and not on the sister division’s compliance with the OEM’s purchasing agreements, specific product requirements, and related activities. Some form of augmentation audit might be needed in addition to the corporate audit.
FDA has also stated that a satisfactory FDA audit of the supplier does not suffice either. Instead, it could be used as a good data point, perhaps as a way for reducing the scope of the OEM’s audit. As with the corporate audit, it does not eliminate the need for a manufacturer-specific audit.
In theory, these supplier controls are overdue—14 years overdue for medical devices, based on the introduction date of the QSR. For FDA, business costs are not part of the equation. For the manufacturer, cost is an important factor. At the end of the day, the manufacturer is ultimately responsible.
The reality is that it will take the industry years to come up to speed on the latest cGMP requirements. FDA and global regulatory bodies have raised the bar on supplier controls through recent enforcement actions and issuances of guidances, especially in the area of supplier process validation.
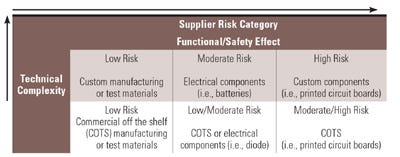
Supplier Risk Grid
|
Figure 1. The risk management process should include a supplier risk grid that identifies device categories and the level of risk. |
Manufacturers must identify high-risk component suppliers through a documented risk management process. Typically, design and process failure mode and effects analysis call out the function and effects of failure of the component design or process output and detection controls. Other more efficient methods can be used, such as component category risk grid methodologies that prioritize remediation efforts based on the degree of customization and effect on function and safety (see Figure I).
Manufacturers should develop a quality plan with a realistic timeline to address high-risk (i.e., 6 to 12 months) and moderate-risk (i.e., 12 to 24 months) components with an approach on how to address low-risk components in the long term. They should identify any additional interim incoming supply quality controls needed until remediation is complete. In the end, manufacturers must balance risk, cost, and quality while maintaining regulatory compliance.
Manufacturer Challenges and Solutions
The challenges presented by the stricter interpretation and enforcement of the supplier control regulations are increasingly dynamic for established firms with long-standing supplier relationships and historical paradigms and practices to overcome. These challenges include ensuring that the established quality system elements continue to evolve to meet the stricter interpretation of the regulations around supplier controls.
The continued improvement of established supplier controls includes extending the detailed monitoring of supplier production and process control parameters and ensuring that the supplied part realization processes are validated to the guidelines established by the GHTF. Strategies such as using production part approval process (PPAP) are compatible with the GMP regulations and have been successfully deployed across many industries that rely on robust and consistent supplier performance. It includes elements such as demonstrated and monitored process capability, comprehensive process control plans, identification and verification of critical to quality part characteristics, and comprehensive first article inspections, all of which are standard considerations of a robust PPAP deployment.
To ensure that supplier processes remain qualified to the current standards, a deliberate and targeted approach is necessary. The suppliers that produce parts that present the highest risk to the operation of the finished device are prioritized for first consideration and, when necessary, updated to current standards.
Pending the completion of any required supplier process revalidations, a review of the supplier’s process monitoring data gathered during production can be performed by the manufacturer, as an interim and mitigating control. This process is part of incoming quality assurance (IQA) inspection activities, which ensure that the processes are maintained in their preestablished state of control be the supplied parts are used. Alternatively, the OEM may choose to increase IQA internal test sampling using a tightened inspection plan. These interim control(s) can be discontinued once the revalidation work has been successfully completed.
Following the completion of the revalidations, the monitoring of supplier performance is expanded to include the periodic review of supplier production quality data. This data is reviewed on a routine basis to ensure that the suppliers are maintaining their established levels of process capability and that their internal controls are functioning effectively.
Partnering with each supplier in the gemba, a Japanese term used in connection with the lean Six Sigma manufacturing concept that means “out on the floor,” ensures that the process validations are successful. In this case, the gemba is each supplier’s respective factory floor, which is where the manufacturer and the supplier work together. The development of the plans and protocols is a joint activity, and approvals from both firms are required prior to execution. The validations include standard installation qualification, operational qualification, and performance qualification considerations. They incorporate standard statistical rationale for sampling and confidence and reliability intervals. The burden of cost associated with these revalidations to the current standards should be shared between the firm and the suppliers, as the benefit is mutual and the requirement common to the medical device industry.
The benefits of institutionalizing increasingly rigorous supplier controls go well beyond sustained regulatory compliance. They extend into tangible business outcomes by improving customer intimacy through fewer supply chain interruptions of commitments and increasing operational leverage by lowering the overall cost of quality.
Supplier Challenges and Solutions
Device manufacturer quality system purchasing controls requirements are creating unique challenges for suppliers. Key challenges include
? Balancing the role of being both a customer and a supplier. In many cases, the supplier to the final device manufacturer isn’t last in the supply chain. Suppliers may also receive critical components and supplies from what would be considered subtier suppliers. Since they aren’t the final device manufacturers, they may not be in a position to know which of the subtier processes and components are critical.
? As OEMs react to FDA’s expectations, many are hastily implementing new controls, which may vary significantly from one device manufacturer to another. These variations in requirements can manifest themselves into significantly different internal requirements for suppliers, which at a minimum, can add costs to internal processes and create confusion for staff.
? Significant new control requirements must come at a cost, and the pressure frequently falls on suppliers to absorb these costs. Although the increased controls will ultimately benefit both suppliers and device manufacturers, the implementation of costs must be shared.
A practical way to resolve these challenges is to develop, approve, and implement a common supply quality agreement (SQA). The SQAs initiate from a common template for all customers and enable suppliers to establish a common framework that takes into account the applicable international (ISO 13485) and U.S. regulatory (QSR) requirements. The common template is customized to take into account each unique customer requirement, including references to the manufacturer’s QSR.
Once completed, the SQA establishes clear definitions of supplier and customer (the device manufacturer) responsibilities. Key topics covered by the SQA are
? Ownership of product specifications.
? Inspection plans.
? Audit functions.
? Complaint handling processes and responsibilities.
? Change control.
? Process validation.
? Process controls.
? Design controls.
? Control of subtier suppliers.
? Legal aspects.
? Key personnel responsibilities and contact information.
Although these elements are critical to the success of the device manufacturer-supplier relationship, one of the most challenging elements is change control. The challenge frequently comes from poorly worded agreements, which can include language that refers to “significant changes” or “changes that could affect the finished device.” In many cases, the supplier is asked to make changes that originate in its processes, but which they might not be fully qualified to make. For example, a change that may have a minimal effect on a supplier’s process may affect regulatory notifications. Ensuring that effective change control processes are fully implemented requires
? Clear language within the SQAs.
? Effective supplier quality system change control processes.
? Consistent provision of change control information to the manufacturer.
? Oversight by the supplier’s and manufacturer’s audit processes.
Another important element of design control requirements is design transfer. This is always a critical phase for suppliers because once the customer receives FDA clearance to market a device, there is almost always a significant push to move quickly from a preproduction or development environment to full production. This is another example where the SQA adds significant value to the process. A typical agreement may require the use of identical equipment. This may be very expensive, if not impossible, to achieve. In those cases, the SQA defines, upfront, the flexibility that a supplier has in meeting the increased production demand as well as all quality and regulatory requirements.
Process controls must be implemented and demonstrated to the customers by both classic verification and validation where required by the QSR. The initial cost of validating a process can be significant, but doing so inevitably adds value by ensuring a stable process that consistently achieves desired results with a high degree of assurance. Once a validated state is achieved, process controls must be established to ensure processes remain in a state of control.
A special cost-saving validation method is called a common validation. For example, complete part families are grouped within a single validation by using at least a two-corner validation approach, which covers a range of parameters for one or more processes. Future parts that are similar to already validated parts do not require additional process validation because they are already covered by the common validation of these processes.
It is important to build and maintain internal centralized validation knowledge to ensure actuality and enhancement of common validations regarding new products and enhanced process parameters.
Another important topic for suppliers and device manufacturers is remediation. All the practices outlined in this article are challenging enough to implement on a go-forward basis, but retrospective application can be daunting. Although there is not a one-size-fits-all solution here, some key considerations should include the following:
? Under whose quality system requirements will the work be performed? As part of the process of becoming an approved supplier, suppliers must demonstrate that their quality system processes meet all applicable international quality systems, U.S. regulations, and device manufacturer requirements. All work should be performed under the requirements of the supplier’s QSR. The extent of the oversight (review and approval) by a supplier’s customer should be defined by the SQA.
? When resources are provided by the manufacturer, who is responsible for managing the overall resource effort? Since the work is being performed under the requirements of the supplier’s quality system, and in most cases, the work is being performed at the supplier’s site, it is recommended that the supplier manage the effort. As with most resources, the cost of managing the project should be shared.
? Who will fund the retrospective review and implementation activities? There is no simple answer here. Because there is a shared benefit of bringing older processes in line with current requirements, these costs should also be shared. There are other ways to resolve cost situations, such as negotiating additional product volumes.
Conclusion
As FDA focuses on purchasing controls, the following is clear
? FDA does not see this as a new requirement. It expects OEMs to conduct remediation (where required) within aggressive timelines.
? The bar is rising quickly, in particular for device manufacturers, as they comply with FDA’s expectations. Device manufacturers that do not take the issue seriously are likely to face regulatory action.
? Where remediation is required, it is appropriate to use a risk management approach to prioritize work but not to eliminate it. Manufacturers cannot risk manage away regulatory requirements.
? A detailed SQA that accounts for the OEM’s QSR and clearly defines the OEM’s and supplier’s area of responsibility should be developed.
? Suppliers should not necessarily wait for direction from their OEM customers in this area. They need to look inwardly at their quality system processes to ensure that they are meeting the intent and spirit of the regulations. If they have not heard from their customers, they should begin reaching out to them immediately to ensure their combined processes are aligned.
? Collaboration between manufacturers and suppliers is critical to success. Both manufacturers and suppliers share the risk and will also share in the benefit when processes are properly validated and appropriate process controls are established.
The time to act is now. Depending upon the number of products that a manufacturer has on the market, the complexity of the processes that are outsourced, and the depth of the supply chain, the effort required to come into full compliance can be significant. However, the effort is well worth it in the long term. Manufacturers and suppliers that heed regulatory compliance avoid regulatory action, improve business outcomes, and most importantly, decrease patient safety issues.
References
1. J Avellanet, “FDA Drug Enforcement: An Analysis of Warning Letter Trends,” FDANews, March 2010.
2. Seventh Annual Medical Device Quality Congress, Bethesda, MD, June 2–4, 2010.
3. FDA, Medical Devices: Current Good Manufacturing Practice (cGMP) Final Rule; QSR, Federal Register, 61 (195), Supplementary Information, Section V. Response to Comments and Rationale for Changes, comments no. 7, 99, 106, 107, and 111, October 7, 1996.
4. Quality Management System–Medical Devices–Guidance on the Control of Products and Services Obtained from Suppliers, GHTF/SG3/N17:2008, GHTF Study Group 3, December 11, 2008.
5. K Trautman, "The GHTF Document on Supplier Controls," FDA, CDRH, Seventh Annual Medical Device Quality Congress, Bethesda, MD), June 3, 2010.
Braulio Ortiz is cofounder and project manager of BioTeknica Inc. (Miami). Michael Neaves is senior quality consultant of the company. Jim Dabbs is vice president of quality management and quality assurance of Physio-Control Inc. (Redmond, WA). Peter Stein is director of quality assurance at Lohmann & Rauscher International GmbH & Company KG (Neuwied, Germany).
About the Author(s)
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
