

Sign up for the QMED & MD+DI Daily newsletter.
Biocompatibility Testing: Tips for Avoiding Pitfalls, Part 2
According to FDA regulations, certain medical devices need to be evaluated for biocompatibility. As a start, manufacturers may search FDA’s Web site to see what comes up on the topic of biocompatibility testing. Many search results will include references to documents such as ISO 10993 and the Blue Book Memorandum No. G95-1 (“Use of International Standard ISO 10993”).
Laurence Lister
February 1, 2010
16 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Biocompatibility Testing: Tips for Avoiding Pitfalls, Part 2")
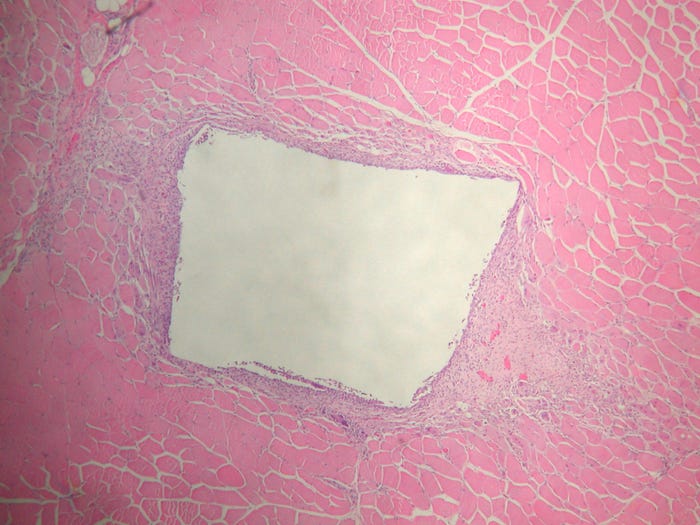
|
Intramuscular implantation assesses local tissue response to an implanted material for the ISO 10996-6 implantation test. |
However, manufacturers are on their own when it comes to how best to fulfill the testing requirements. This article is a continuation of “Biocompatibility Testing: Tips for Avoiding Pitfalls, Part 1,” which appeared in the January 2010 issue of MD+DI. It discusses several more elements of biocompatibility testing and also explores how to avoid problems that may arise along the way.
Cytotoxicity Testing
Cytotoxicity testing may be considered the canary in the coal mine of biocompatibility testing. Three basic categories of testing are required for every medical device, no matter how simple it may be. These categories are cytotoxicity, sensitization, and irritation and intracutaneous reactivity. The latter two are animal tests, and consequently are more expensive and take longer to get results. Therefore, when testing novel materials, the cytotoxicity testing is typically performed first. This serves as a barometer as to how the rest of the testing is likely to proceed, and sponsors with questionable materials advance only after the results of cytotoxicity testing are known. Some material vendors may only perform cytotoxicity testing before claiming that their product is a medical-grade material. Manufacturers should insist on certificates (test reports) for any material they purchase that is claimed to be medical grade.
The purpose of cytotoxicity testing is to determine the biological reactivity of the mammalian cell lines following contact with the material or the extract of the material. Cells are grown on plates. Depending on the test being performed, the cells are either derived from the ovaries of Chinese hamsters or from fibroblasts of mice. Once the cells reach an appropriate confluency or density, the test article, or extract of the test article, is exposed to cell population. Following the exposure, there is a period in which the plates will be observed for apparent cell death.
Cells are very sensitive and can be killed by minor changes in pH, salinity, or temperature. It is possible to have some incidence of this type of cell death, and this does not classify the test article as cytotoxic. But if the vast majority of the cells are dead or dying, the test article will be classified as cytotoxic.
|
Both in vitro and in vivo tests are required as part of a testing battery. |
Sensitization
Sensitization testing establishes the potential of the test article to elicit an allergenic response. In particular, the testing is aimed at a delayed-type hypersensitivity response. This is an immune response that takes a couple of days to develop. Poison ivy is a good example of a delayed-type hypersensitivity response.
Two primary test methods are used for medical devices to satisfy the sensitization testing requirement. The first is the guinea pig maximization test (GPMT); the second is the closed patch test, also known as the Buehler. Due to the use of an adjuvant to stimulate the immune system, the GPMT has been considered the more sensitive test. The GPMT has intradermal injections in the beginning of the study and topical applications at the conclusion. The Buehler consists of all topical applications. Due to the intradermal injections in the GPMT, the requirements specify that the test article be a liquid, suspendable powder, or extract. The Buehler is better performed with surface contact devices as they are used, if possible, and chemicals.
The third commonly used sensitization test method is called the mouse local lymph node assay (LLNA). The LLNA is an acceptable test method, but device manufacturers have been using the guinea pig tests for decades and are satisfied with the results obtained using those methods. The LLNA, like any test, has its pros and cons. It is shorter in duration, uses animals of a lower phylum, and requires far less test material; those factors can be appealing. On the other hand, it generates radioactive waste and is not accurate in distinguishing a sensitization response from an irritation response, which increases the possibility of a false positive. In addition, because the animals are sacrificed to harvest the lymph nodes, there is no opportunity to rechallenge if a question arises.
When proceeding with any sensitization study, it is necessary to know the irritancy potential of the test article. As already mentioned, the LLNA may confuse irritancy with sensitization. In the two guinea pig tests, the animals are scored for sensitization by evaluating erythema and edema (redness and swelling). Coincidentally, redness and swelling are also indicators of irritation. So it may appear that the GPMT and the Buehler could give a false positive to an irritating test article. But these studies have safeguards to prevent this. They are designed with primary irritation animals that receive varying concentrations of the test article all on one day at the same time. Because the animals have not been exposed to the test article before this point, any redness or swelling is considered an irritation response and not an allergenic response. These results are then considered when preparing the dosing solutions for the main part of the study. At the challenge dose, which is the final determining dosing, the animals are dosed at the highest concentration determined to not produce any irritation; any observation noted will be considered an allergenic response.
Irritation
Irritation testing determines whether the test article will cause irritation to the appropriate tissue. Various models are available to test an article. The most common model is referred to as the intracutaneous reactivity study (IC). In this test, extracts are typically used to inject five 0.2-ml boluses per extract into the skin of rabbits. The injections are usually the size of a mosquito bite. The skin sample area is observed every 24 hours for 72 hours following the injections and is scored for erythema and edema. The test sites are compared with control sites, and in order for the test article to pass, the difference of the average scores cannot exceed 1. The IC test can also be used with the test article directly if it is small enough to pass through a needle.
Other irritation models involve ocular, mucosal (oral, penile, bladder, rectal, and vaginal), and dermal tissues. The irritation test should use the most appropriate tissue for that device. For example, if the product is a contact lens case, then ocular irritation should be performed. A vaginal speculum should use a vaginal irritation study.
Even if appropriate tissue irritation is being performed, such as the ocular irritation in the instance of the contact lens case, consideration should be given to performing an IC test as well. On a topical dermal study, the test article is in contact with the epidermis. The epidermis is the first line of defense to protect us from the environment, and it has evolved to be fairly effective for that purpose. Therefore, some materials may not be detected as irritants by a topical dermal exposure, even if that is the most likely route of human exposure. Oral, rectal, vaginal, penile, bladder, and ocular tissues all have the ability to expel the test article or extract. The oral irritation study can utilize hamsters fitted with retaining collars to prevent a solid test article from being expelled. With the IC, the test article or extract is embedded within the dermis, and provides another evaluation as to the potential irritancy of the test article.
|
Shown here are mouse fibroblast L929 cells in culture for the ISO 10993-5 cytotoxicity assay. |
Systemic Toxicity
In discussing systemic toxicity, one must first understand that the term systemic refers to the way the animal is affected as whole. In systemic toxicity testing, the animal is exposed to the test article or the extract of the test article. Four categories of systemic toxicity testing exist, and each is broken up by duration. The first is called acute systemic toxicity. This is a single exposure with typically a 72-hour observation. The next category is called subacute, which is a misnomer. In subacute toxicity testing, the animals are exposed to the test article or extract for 14–28 days.
Subchronic toxicity testing is a 28–90-day exposure, and chronic toxicity testing is a 6–12-month exposure. In the toxicity studies discussed in this section, the shorter duration is indicated when the test is conducted using an intravenous administration. The longer duration is for all other routes of administration. The category of testing required should be similar to the clinical exposure. Most medical devices will not need to pursue chronic toxicity testing. As the exposure period increases, so does the number of animals required. In these continuous or repeat dose studies, the animals are necropsied at the end of the study, and pathological evaluation is performed on the organs of the animals. In addition, chemical, coagulation, and hematological evaluations are performed on the blood.
The fourth type of testing that falls under the systemic toxicity category is called pyrogenicity testing. It assesses whether or not a test article has the ability to cause a feverlike response. In the United States, there are currently two types of tests for pyrogenicity, one is in vitro and the other is in vivo. The human monocyte test has been approved in Europe as an alternative to the rabbit pyrogen test. The in vitro test, known as either the bacterial endotoxin test or Limulus amebocyte lysate (LAL), only detects pyrogens that are bacterial in origin, called endotoxins. The in vivo test is called the rabbit pyrogenicity test; this detects bacterial endotoxins as well as material-mediated pyrogens. Therefore, the rabbit pyrogenicity test is required during the initial evaluation of the product, and the bacterial endotoxin test is used as a surveillance tool for bacterial con-tamination during manufacturing.
LAL uses a component of the blood of horseshoe crabs. In the presence of endotoxins, a component in bacterial cell walls, the LAL will clot. Using standard curves, the amount of endotoxin present is quantifiable. The rabbit pyrogenicity test involves monitoring the temperatures of rabbits for three hours following an intravenous injection of the test article or the saline extract of the test article. If any rabbit’s temperature increases half a degree Celsius following the injection, the test is considered positive.
Pyrogenicity testing is not to be confused with sterility testing. In the sterilization process, the bacteria on a material are killed but not removed. When the body is exposed to these dead bacterial components, it recognizes the presence of bacteria and responds with a fever as if there were an active infection. So just because something is sterile doesn’t necessarily mean it is nonpyrogenic.
Genotoxicity
Genotoxicity testing evaluates the test article’s ability to cause damage to DNA, genes, and chromosomes. Because of this, genotoxicity testing is performed as a battery. Genotoxicity testing typically consists of the bacterial reverse mutation assay, also known as the Ames assay, the mouse micronucleus test, and the mouse lymphoma or chromosomal aberration test.
The Ames assay uses bacteria that have been mutated to require specific amino acids in order to survive. The bacteria are exposed to the test article in the presence of just enough of these essential amino acids to allow for a limited number of replications. A proliferation of bacteria’s DNA indicates that the bacteria reverse mutated back to wild type bacteria and that these limited amino acids were no longer essential to survive.
The mouse micronucleus test is an in vivo test in which mice are exposed to the test article or extract. Bone marrow is harvested from the animals and is evaluated for the presence of micronuclei. Micronuclei are comprised of chromosomes or fragments of chromosomes and are indicative of chromosomal damage.
The mouse lymphoma test uses a mutated mouse cancer cell line in which a partially damaged gene exists. When this gene is completely damaged, this mutated cell line is able to survive and replicate in the presence of a particular chemical. The cells are incubated within that chemical after exposure to the test article. If an increase in viability is detected, this indicates the test article was able to totally inactivate or damage the gene.
The chromosomal aberration study typically uses cells derived from Chinese hamster ovaries (CHO cells). These cells are encouraged to undergo mitosis, or cell division. They are then exposed to the test article and a chemical that stops the mitosis in metaphase stage of mitosis. This is the stage in which all chromosomes are visible. At least 200 metaphase cells will be evaluated for visible damage to the chromosomes.
Genotoxicity testing is a crucial parameter of this area of testing. It is required for anything that will come in contact with the body for over 30 days and anything that enters the body for more than 24 hours. Genotoxicity testing is important because, if something can cause genetic damage in these tests, it has the possibility to cause cancer.
Implantation
In implantation testing, the test material is placed into the body of test animals. Various species can be used for the implant test, ranging from mice to swine. The most typical animal used is the rabbit. Due to the size of the rabbit and the quantity of sites required to fulfill the guidelines, the rabbit is the most suitable model. Switching to rats is likely to increase the number of animals required to obtain the minimum amount of sites. Using more animals increases the cost.
The tissue chosen for implantation should be tissue that is most suitable for the test article. When in doubt, perform a muscle implant test. One benefit is that with the muscle implant, the test article is contained in a well-vascularized tissue, and this affords the body the maximum opportunity to react with the test article. For instance, a vascular shunt should be tested with a muscle implant test. In actual use, this device will be sutured into a vessel. Logic dictates that that test article be placed in an appropriate vessel in an animal, but this process should be for efficacy testing, not biocompatibility testing.
By using this device in the same manner as clinical use, the animal will need to be treated the same as potential patients. Anticoagulants and antibiotics will be administered to the animals, just as they would be with patients. Also, the surgical process is severe—sutures are introduced, cautery is used, and trauma as a result of clamping is created during the procedure. These variables prevent getting a true profile of how a body may react to this material. The best way to capture a potential reaction is to place small pieces of the test material into discrete pockets in the muscle along the spine of rabbits. That particular location is a single muscle mass, so there is minimal movement of the material after implantation.
Other sites for implantation would be into the subcutaneous space; bone, ocular, dental, or mucosal sites; or into the brain. If one of these locations is more appropriate for the finished test article, then that would be the better choice; otherwise use a muscle implant for your biocompatibility evaluations.
The duration of these implant studies is dictated by the clinical exposure time of the test article. Multiple time points will need to be conducted. For a permanent implant, a minimum short term of 1–4 weeks should be performed and a long term of over 12 weeks. Also consider doing a midterm of about 8 weeks. The point is to establish a steady state of the test site. In other words, if something from the test article was leaching out, it would be noticed in the short-term implant, but may have healed by 13 weeks. Or if something took a while to start leaching or degrading, nothing may be observable in the 1–4-week time point, but it will be evident at 13 weeks. Particular consideration should be given to the time point selection when testing resorbable or biodegradable materials. These selections should be based on the degradation rate of the test material.
At the end of the study, the implanted sites are retrieved and processed histopathologically. A pathologist evaluates the sites and scores them for a local tissue reaction. The scores from the control sites, which will be implanted with a proven biocompatible material, are subtracted from the scores of the test sites. This final score then categorizes the test material into a gradation of irritancy.
Hemocompatibility
Hemocompatibility testing includes a full battery of tests required of anything that comes into contact with blood. ISO 10993-4 clearly defines the categories of evaluations to be performed, as well as which types of medical devices need to have which tests performed. The five categories of hemocompatibility testing are thrombosis, coagulation, platelets, hematology, and complement system. With the exception of thrombosis, all of these tests are in vitro assays.
In the thrombosis study, the test article is implanted into the vasculature of an animal. After a given period of time, the implanted vessel is removed and the test article is observed for clot formations.
The four remaining tests are carried out in test tubes and evaluate specific actions: whether or not the test article has an effect on the blood’s ability to clot or coagulate, can regulate an immune response, or can damage the cellular components of the blood.
Supplemental Testing
Supplemental testing involves chronic toxicity testing and reproductive toxicity testing, as well as carcinogenicity studies and degradation studies. These are all long tests with high price tags. If such testing is required, consult qualified individuals to assist; a board-certified toxicologist or other professional with the proper education, training, and experience would be invaluable. These supplemental studies are not as cut and dried as the tests discussed earlier in this article. It’s advantageous to consult with someone who can put together appropriate testing for the product.
Conclusion
Medical device biocompatibility testing is filled with potential pitfalls in areas such as genotoxicity, irritation, and implantation. Because these pitfalls could delay a product launch, manufacturers must follow strict testing protocol and begin the testing process early enough to allow for thorough and complete testing. Manufacturers should also be as up to date as possible on all requirements, and they should consult other qualified professionals to provide expertise.
Bibliography
“Biological Evaluation of Medical Devices,” ISO 10993, parts 1–12 (Geneva: International Organization for Standardization, various dates).
“Use of International Standard ISO 10993, Biological Evaluation of Medical Devices—Part 1: Evaluation and Testing” G95-1 (Rockville, MD: Department of Health and Human Services, FDA, 1995).
“Testing Methods to Evaluate Biological Safety of Medical Devices, Notice from the Office Medical Devices Evaluation Number 36” (Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare, March 19, 2003).
“<87>Biological Reactivity Tests In Vivo.” United States Pharmacopoeia, most recent version.
“<88>Biological Reactivity Tests In Vitro.” United States Pharmacopoeia, most recent version.
“<1031> The Biocompatibility of Materials Used in Drug Containers, Medical Devices and Implants.” United States Pharmacopoeia, most recent version.
“Standard Practice for Selecting Generic Biological Test Methods for Materials and Devices.” ASTM F748-06.
Laurence Lister is director of biocompatibility services at Toxikon Corp. (Bedford, MA).
About the Author(s)
You May Also Like


