






Sign up for the QMED & MD+DI Daily newsletter.
Common Mistakes in Validating Package Systems
Originally Published MDDI May 2006 TESTINGSome common pitfalls can occur during package development and validation. Here’s how to avoid them. Patrick J. Nolan
Patrick J. Nolan
May 1, 2006
19 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Common Mistakes in Validating Package Systems")
TESTING
A medical device's package plays a key role in safely delivering treatment to patients. It must ensure the integrity of the device from the point of manufacture to the point of final use. In addition, packaging often has a direct function in the application of the treatment; for example, it may act as a fixture or a dispenser. Therefore, mechanical damage to a package cannot be tolerated. Device components and packaging systems must combine to create a product that performs efficiently, safely, and effectively in the hands of the user.
The packaging process is extremely important as well. Regulatory authorities recognize the critical nature of a sterile barrier system. In fact, they consider packaging an accessory or a component of the medical device, which implies that the package system is nearly as important as the device itself. The package must keep a device sterile throughout all the stresses and hazards generated by the manufacturing, shipping, and storage environments. Ultimately, any device that is labeled as sterile but arrives nonsterile at the point of use can critically compromise patient safety.
The design and development of packaging systems have come under scrutiny by both international and domestic regulatory agencies. This scrutiny has placed a great deal of emphasis on standardizing the package development process. Some standardization has come from the International Organization for Standardization in its standard, ISO 11607.1 Although the existing standards provide guidance, there are some common pitfalls that occur when developing and validating a package system for a terminally sterilized medical device. It is important to know what they are and how to avoid them.
|
Figure 1. The sharp radius of the product caused a cut in the poly component of this package. |
Sterility Is Paramount
The most common defect resulting from subjecting packaged devices to manufacturing, sterilization, handling, and storage is loss of sterile integrity. Pinholes, slits, cuts, and tears of pouch packages, as well as fractured thermoforms, can all compromise sterility (see Figures 1 and 2). These defects occur from impacts caused by dropping packages and from general handling or mishandling. Vibration forces generated by the movement of trucks over the road and aircraft in flight can also cause defects.
|
Figure 2. Creasing and wrinkling a package can result in pinholes in the plastic film. |
The manufacturing and assembly process can cause some defects, including tears. For example, any sharp edges or staples may snag the packaging materials when placing pouches or inserting information booklets into cartons. Device makers can minimize package defects caused by handling and shipping by designing sterile barrier and package systems that reduce the possibility of pinholes, tears, and fracturing.
Perform Adequate Testing
Many device firms are not aware that they need to test package systems. Some are not even aware that ISO 11607 exists and is used by FDA and the European Community to ensure that package validation activities were conducted properly. In addition, many companies cut corners by trying to complete package validation without using recognized scientific practices.
In an attempt to get their product to market first, some device makers do not allow enough time in the product development process to perform adequate package system validation. Alternatively, they cut corners in the process and thus risk regulation violations or recalls. Ultimately, cutting corners could allow suspect devices to reach the market and the patient.
But when should device makers perform a full package system validation, including package design, manufacturing process qualification, and protective package performance qualification? It depends on the expected shelf life of the product and the desired expiration dates. The typical time frame from package concept to final qualification for a one-year shelf life could be three to six months.
Also, extra time should be allowed for any unexpected events in the validation. The validation process path should parallel the product development process. This parallel path can begin when the basic concept and model of the product is determined. One strategy is to use prototype products for design elements as well as package compatibility and testing requirements. If a longer shelf life is desired, the process time line must be extended by about 45 days for each year of shelf life. The bottom line is that the package development process cannot be ignored, and it must be done correctly.
Prequalify the Package-Product Compatibility
It is essential to do some preliminary design evaluation work before diving into the package system validation. Cutting corners to save time and money usually results in extended development times and higher overall validation costs because some attribute of the package system often fails during validation. And, any failure during validation requires retesting. Any failed package system attributes would likely have been detected in preliminary testing
projects prior to validation if the proper work had been done up front.
Seal strength and seal integrity tests are common prequalification tests that should be used to detect potential design or manufacturing problems. These tests identify any deficiencies in the manufacturing process and indicate that corrective action is necessary on the production line. The tests should be done far in advance of any package performance tests (e.g., transportation, sterilization, handling, etc.). They also should be used as a basis for establishing target values for process quality control. Tests such as ASTM F88 for seal strength and ASTM F2096 for package integrity are commonly used to evaluate sterile barrier systems.2,3
Another way to prequalify package-product compatibility is to perform testing associated with transportation and handling. Common laboratory tests used to simulate the forces inherent in the distribution and handling environment include ASTM D4169 and International Safe Transit Association (ISTA) preshipment tests. It is accepted fact that sterile medical device packages do not lose their sterility simply by being stored on a shelf. Therefore, most package failures result from the dynamics inherent in the handling and distribution environments. In essence, package failures can result from events that may occur during manufacturing, shipping and handling to and from the sterilization facility, or in distribution. Each process can subject the finished package to shock, vibration, changes in atmospheric pressure, and high and low temperature extremes. With so many potential places for failures, proposed package designs must be put through prequalification processes. Each specific test can isolate potential hazards to determine the package-product response and compatibility.
Identify Worst-Case Scenarios
|
Figure 3. Many protective packages or shipping boxes may not be filled completely, causing void spaces that must be filled with paper or plastic cushioning materials. |
It is often difficult to determine which shipping unit to actually test. Many device makers do not configure their shipments in homogeneous products and quantities (e.g., the same product in a 24 pack). Rather, they vary the products they ship and often have different quantities for every shipment. Because of this inconsistency, it is hard to perform a test that represents a typical shipment. It is important to establish the most common configuration for shipping the product prior to package validation and to account for the worst-case scenario (see Figures 3 and 4). The worst-case package configuration can be determined by a prequalification test performed on various configurations. Once a worst-case scenario has been validated, any other package configuration of the product can be covered by that validation. Only a minimal amount of testing may be needed on other package configurations, rather than a complete validation. Another benefit of forethought in this aspect of package development is that firms may be able to invoke a provision in ISO 11607. That particular provision allows families of packaged products, rather than each individual package configuration, to be validated.
|
Figure 4. The sterile barrier system is bulk-packed in this protective package using void-space fillers such as EPS peanuts. |
Perform IQ, OQ, and PQ
The notion of validating package forming and sealing equipment will gain more scrutiny and emphasis in the future owing to the revisions taking place to ISO 11607. In the quality system regulation, FDA has mandated validation for processes that cannot be completely verified, such as package integrity. However, it is not uncommon for device manufacturers to operate packaging equipment based solely on a vendor's recommendations. Manufacturers should perform formal installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) on their package design and materials under their standard operating procedures.
One requirement in the revised ISO 11607 mandates that packages be produced at the low end of the process parameter range for performance testing. In addition to ensuring a controlled process and producing higher quality packages, the requirement makes it mandatory to perform a formal IQ, OQ, and PQ validation.
The primary objective of a package process validation should be to provide a high degree of assurance that products will reach users in a condition to be used properly and as intended. FDA provides the following definition for validation in its Guideline on General Principles of Process Validation:4…establishing by objective evidence that the process, under anticipated conditions, including worst case conditions, consistently produces a product which meets all predetermined requirements (and specifications).
In a practical sense, a process validation must address the requirements or applications of the package, the interaction of people and equipment during its manufacture, the consistency with which it can be made, the effects of processing on its performance, and its storage and handling. Manufacturers must become intimately involved with how products are packaged and how to maintain their consistency and uniformity. In addition, they must have proof that such processes perform as they were intended.
There are many specific benefits of package process validation. It reduces the risk of product malfunction and the potential for a nonsterile operating condition. It can also improve customer satisfaction, improve manufacturing efficiencies, reduce costs, and reduce development times. Of course, compliance with regulatory requirements is also an important benefit. The FDA guideline provides valuable information about quality system requirements.
|
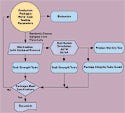
Figure 5. One possible scenario for completing a package process validation is shown in this flow chart. |
Primarily, validation establishes the process parameters that produce acceptable package performance. Validation may be performed by testing packages produced from a matrix of process-parameter variable combinations. In some cases, a design of experiments that only tests packages produced at the extreme ranges of the process parameters may be used. Figure 5 shows one course of action for establishing machine process parameters.
Develop a Project Protocol
Prior to starting any work on a validation, it is essential to write a protocol. The protocol provides a blueprint stating how to conduct testing, including the purpose, scope, responsibilities, test parameters, production equipment and settings, and acceptance criteria for the test. Validation requires careful planning and preparation, and it begins with a well-conceived and well-written protocol.
A validation process consists of a series of qualifications of unique processes that make up the complete package process system. This total package process system includes the final package design, the materials used in the package, and the ability to sterilize the product inside its package. The package design and its interactions with the product, the machinery used to assemble the package, the setup and maintenance of the machine, and the consistency of production are other important considerations. If one of these processes is not right, the entire system may break down, leaving the manufacturer at risk of causing harm to patients.
Choose a Suitable Sample Size
Determining the appropriate sample size to use for testing is often daunting. Many factors weigh into the decision, including the type of test (e.g., quantitative variables or qualitative attributes), the sample population, the number of samples available for testing, and the risk factors (e.g., confidence intervals). The most common mistake is choosing a sample size that is too small and that renders results with no statistical significance.
Statistically justifying the sample size can be difficult. This is especially true for tests that produce attribute data (e.g., pass-fail). Variable data are more manageable, because classical statistical analyses such as mean and standard deviation can be used to determine reliable sample sizes. Because device lot sizes are often undetermined, acceptable quality level (AQL) sampling plans cannot be used for package design validation.
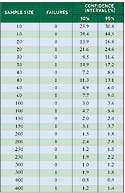
Manufacturers can use reliability statistics to provide an acceptable confidence interval. For data that produce an attribute result, the minimum confidence level must be 95% but may be as high as 98%. This level is usually established by corporate risk management or quality assurance departments. The acceptance criterion for passing this type of test is 0 failures. So, for attribute data, manufacturers can ultimately make a statement, based on their sample size. Such a statement might read, “The true failure rate of the sample at a 95% confidence level (CL) and n = x is y%.”
For example, if 0 defects are found in a sample size of n = 100, the true failure rate is 3.6%. Or, if n = 1000 and 0 defects are found, the true failure rate at 95% CL is 0.36%. Finally, if 0 defects are found and n = 30, the true failure rate at a 95% CL is 11.6%. Of course, protocols and test results are more reliable when they are based on a national or international standard for statistical sampling.
|
Table I. True failure rates for test results from various sample sizes, at two confidence intervals. |
Is this sufficient reliability? Unfortunately, there is no quick answer. It depends on the corporate risk policy, type of product, expected distribution environment (i.e., normal or severe), and critical control manufacturing points (i.e., points where damage could possibly occur). Table I shows the upper boundary of the expected failure rate when 0 or 1 failure is observed at various sample sizes.5
Choose the Appropriate Package Type and Material
Choosing the wrong package material or package type are critical errors in package development and validation. In most cases, it is a package-product compatibility problem that could have been avoided if the package had been prequalified early on. For example, if the product mass is too large for the impact resistance of the plastic material, the thermoform trays may fracture (see Figure 6).
|
Figure 6. Large products can puncture thermoforms and fracture flanges upon impact. |
Fracturing during distribution and handling can be avoided in large, massive products by using high-impact-resistant plastics such as polycarbonate. The thermoform design is also critical to ensure that the product is held in place firmly, so that a loose product is not jettisoned through the tray lid or does not fracture the plastic from the inside.
If fracturing cannot be avoided by material selection alone, then provisions for cushioning the thermoform must be considered. Cushioning is most effective at the sterile barrier-system level, where the cushioning is used around the thermoform tray and inside a carton. In addition, cushioning materials may be used at the protective-package, or shipping-unit, level to cushion cartons of products.
|
Figure 7. To allow these pouches to fit into the carton, they were folded, which may cause pinholes at the crease. |
Use Appropriate Carton Sizes
Pinhole defects in pouches can be reduced by inserting a pouch into a carton without folding, wrinkling, or creasing its ends (see Figure 7). Pinholes occur at the junctures of creases and folds when they vibrate. The vibration causes the intersection to be fatigued at the juncture. This effect is exacerbated by complex folds in the pouch, which cause very concentrated points of stress at the junctures of the materials. It is better to use secondary packaging, such as a carton or shelf box, that is large enough to insert the pouch without folding.
Know How to Handle Tyvek Separation
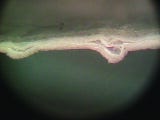
Sheet separation of the porous web of Tyvek is not a new problem. However, it only really came to light when medical device packages began to be integrity tested routinely using bubble and dye leak methods (see Figures 8 and 9).
|
Figure 8. Pouches that are folded to fit into the carton may cause Tyvek to stretch and result in a false-positive leak test. |
Sheet separation can lead to a false-positive result in the integrity test. The false positive occurs when the material is bent, folded, or wrinkled. DuPont has proven that the phenomenon does not change the sterile barrier performance of the material and that any leakage of air or dye solution is only along the transverse direction of the material. (In an adhesive, or seal, failure, the leakage would be between the Tyvek and poly material.) Filtration capability remains when sheet separation occurs. However, when performing bubble and dye leak tests, it is incumbent upon the tester to analyze the failure carefully to determine the type of failure. In cases where there is a suspected false positive, it may be necessary to look at the package under high magnification to determine the cause of the leakage.
|
Figure 9. A magnified view of the Tyvek separation phenomenon. |
Determine Seal Strength Carefully
When a protocol is developed for a package validation, it is sometimes written with restrictive acceptance criteria. One acceptance criterion used for package validations is the so-called standard one-pound seal strength. A minimum seal strength should not be written into a protocol as one of the acceptance criteria. The acceptable range for seal strength should be established during the process validation phase of the package system development. There is no standard minimum seal strength. Rather, the minimum seal strength is that which produces and maintains package integrity. Even if package strength specifications have been established, the package integrity has not necessarily been proven. Package integrity is defined as the unimpaired physical condition of a final package. Seal integrity is defined as the condition of the seal that ensures that it presents a microbial barrier to at least the same extent as the rest of the packaging.”1 Notably, neither definition refers to the strength of the seal. Package integrity is independent of package strength, although a strong package seal indicates a safe package solely from that standpoint. Further, if the entire seal area is homogeneous and continuous, then one could say that the package seal provides integrity. However, this does not address package surfaces that may have undetected pinholes or leaks. Other mechanical tests may be appropriate for determining package seal homogeneity.
Seal strength is important to developing the package process. However, the seal-strength performance specification is used most effectively to monitor the process rather than to determine ultimate acceptance. Seal strength is also an important factor for establishing package process parameters. In fact, ISO 11607 requires that the “seal strength shall be determined at the upper and lower limits of the defined critical sealing process variables and shall be demonstrated to be suitable for the intended purpose.”
Package integrity at the point of final use is the principal acceptance criterion for a sterile medical device package. However, performance attributes are essential to the package design and development process. The critical acceptance criterion for any package validation protocol should be package integrity.
Perform Accelerated-Aging Tests Properly
In ill-conceived attempts to reduce costs and time, some manufacturers accelerate shelf-life or expiration-date studies to unrealistic limits. They often raise the test temperature to a level that causes packages to melt down, warp, or otherwise change uncharacteristically. Temperatures of more than 65°C are indefensible according to the rationale typically used to justify accelerated-aging protocols.
Accelerated aging is performed on packaged medical devices to document expiration dates for products. Real-time aging can be performed; however, many products would be obsolete by the time a three-year expiration date had been validated. FDA does not require expiration dating for products without components and with a defined effective life, such as batteries. The European Directives imply that all sterile medical devices must have an expiration date. Therefore, documented evidence must exist to substantiate claims related to expiration dating.
Accelerated aging is based on a thermodynamic temperature coefficient formulated by Von't Hof. It states that for every 10°C rise in temperature, the rate of chemical reaction will double.6 However, that formula was based on the rate kinetics of a single chemical reaction, and not on packages made from various materials. So directly extrapolating this theory to the aging of packaging materials must be done cautiously. Nevertheless, the theory is useful for defining and justifying accelerated-aging test programs.
The test duration for an accelerated-aging study is determined using the following formulas:
Accelerated Aging Rate (AAR) =
Q10((elevated temperature – ambient temperature)/10)
where Q10 is 2.0 and ambient temperature is 23°C or 16°C, and
Accelerated Aging Time Duration (AATD) =
Desired Real Time Aging/AAR.
Temperature selection for accelerated-aging studies should avoid unrealistic failure conditions, such as deformation from melting.
Conclusion
There are many factors that need to be considered when performing a package validation. First, sterility is the most important consideration. A nonsterile product could have grave effects on end-users. The most basic way to ensure sterility is to perform due diligence. Do not cut corners or skip IQ, OQ, or PQ activities. Manufacturers need to take the time to develop appropriate protocols and to consider package-product compatibility and materials selection. Doing things the right way the first time will result in more-controlled processes and ultimately safer products.
References
1. ISO 11607, “Packaging for Terminally Sterilized Medical Devices” (Geneva: International Organization for Standardization, 2000).
2. ASTM F88-05, “Standard Test Method for Seal Strength of Flexible Barrier Materials” (West Conshohocken, PA: ASTM International, 2005).
3. ASTM International F2096-01, “Standard Test Method for Detecting Gross Leaks in Porous Medical Packaging by Internal Pressurization (Bubble Test).”
4. “Guideline on General Principles of Process Validation,” (Rockville, MD: FDA, 1987).
5. Thom R Nichols and Sheldon Dummer, “Assessing Pass/Fail Testing When There Are No Failures to Assess,” Medical Device & Diagnostic Industry 19, no. 6 (1997): 97–99.
6. Karl J Hemmerich, “General Aging Theory and Simplified Protocol for Accelerated Aging of Medical Devices,” Medical Plastics and Biomaterials 5, no. 4 (1998): 16–23.
Patrick J. Nolan is chief operating officer for DDL Inc. (Eden Prairie, MN). He also serves as chairman for the ASTM committee D-10 on packaging.
Copyright ©2006 Medical Device & Diagnostic Industry
About the Author(s)
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
