

Sign up for the QMED & MD+DI Daily newsletter.
Exploring Postdevelopment Risk Management
Originally Published MDDI May 2004Regulatory Outlook
May 1, 2004
9 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Exploring Postdevelopment Risk Management")
Originally Published MDDI May 2004
Regulatory Outlook
Tony Chan and Edwin Bills
Agile Pharmaceutical and Bilanx Consulting
Regulations require the integration of risk analysis into all processes, including manufacturing. Applying HACCP principles can minimize the introduction of risk factors.
|
Tony Chan |
Postdevelopment risk management activities should be integrated into the quality system process of medical device development. Most manufacturers already include risk analysis activities in their development processes, so the addition of postdevelopment activities should be relatively simple. Postdevelopment risk management strengthens a quality system. By reporting events that are inconsistent with the initial risk analysis, postdevelopment activities improve a manufacturer's early warning of risk issues.
This article presents a view of post-production risk management. It also offers some methods for integrating risk management and corrective and preventive action (CAPA) processes into a company's quality system.
Postproduction
|
Edwin Bills |
The first paradigm must be to think of “postproduction information” as postdevelopment information. This definition more fully characterizes this portion of the risk management process. The ISO 14971 risk management standard is clear in describing the product development portion of the process, but then introduces the ambiguous title of “Post-Production.” The standard's description of option analysis lists the priority of risk control measures in Clause 6.2, subclauses a, b, and c.
The standard successfully focuses on risk control measures during the design of the medical device in subclause a and in the initial phrase of subclause b. The phrase that follows, however, is “or in the manufacturing process,” which fails to sufficiently convey the importance of risk management during the manufacturing part of the device life cycle.
After Clause 6, the standard does not stipulate any production- or manufacturing-related risk control requirements. Further discussion of risk control does not appear again until Clause 9, “Post-Production.”
Unfortunately, the way standards are written sometimes leads to ambiguity. On the surface, it appears that the authors of this standard omitted the manufacturing process altogether, but this was not the intent. Describing this portion of the risk management process as part of postdevelopment clearly includes the manufacturing phases of a product's life cycle. In addition, it is important to note that the scope of the standard encompasses the entire life cycle of a medical device.
Medical Device Manufacturing and HACCP
|
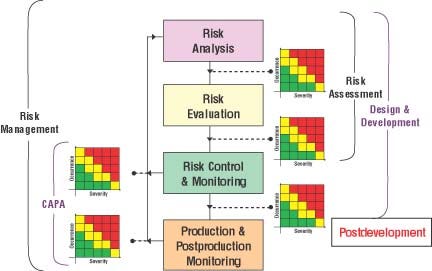
Figure 1. The risk management process (Click to enlarge). |
The manufacturing process for a medical device is an important part of the risk management process, which is illustrated schematically in Figure 1. Product risks can certainly be mitigated during this process.
The hazard analysis and critical control points (HACCP) technique was implemented initially by FDA's Center for Food Safety. Prior to the adoption of the quality system inspection technique (QSIT) in 2000, FDA promoted the use of HACCP in medical device manufacturing. HACCP includes seven risk management steps that can be taken in the manufacturing and preuse part of the product life cycle. Risks can be mitigated by controlling the manufacturing process for risks identified in a risk analysis and by ensuring that measures selected in the implementation section of the risk control phase are controlled.
|
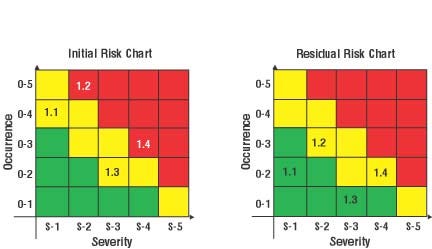
Figure 2. Risk charts for comparing improvement at various stages of product development. The numbers (1.2, etc.) in the matrices are risk identifications for a particular risk item. The same risk chart could be used to trace risk items for other stages in the device life cycle (Click to enlarge). |
Following the manufacturing phase, a manufacturer or others may warehouse, distribute, or install a device. These activities are also candidates for the critical control points identified in the risk analysis, and may require monitoring to ensure that the control levels are not exceeded. HACCP consists of seven steps:
•Conduct hazard analysis and identify preventive measures.
•Determine the critical control points.
•Establish control limits.
•Monitor each critical control point.
•Establish corrective actions.
•Establish verification procedures.
•Establish recordkeeping and documentation procedures.
These seven principles easily work with the risk management process. For example, the first three steps equate to the risk assessment and risk control portions of ISO 14971. The HACCP process is integrated into the manufacturing process so that adequate control measures are taken to prevent the introduction of risk factors into a product during manufacturing. The control measures are also extended into the storage, distribution, and installation processes for the device. Applying HACCP to storage might include measures to prevent damage to the container of sterilized devices. It can also include measures for the removal of expired devices from distribution.
Risk Management and Corrective and Preventive Action Processes
Many quality system engineers have interpreted the postproduction phase of the risk management process as referring to information received from customers concerning the use of the device. This is partially correct, in that postproduction actually refers to all information received from all sources concerning the product. This would include information received from manufacturing, product storage, distribution, installation sources, and product users.
The following paragraphs describe a method for using the information gathered in the postdevelopment phase within a risk management process. It is important to note that the risk management process is an integral part of a manufacturer's quality system and not separate from that system.
When an initial risk analysis for a product is performed during the development process, a risk chart can be created that identifies each risk, its frequency of occurrence, and the severity of its consequences. If risk control measures are implemented for risk, the frequency can be reduced on the risk chart. If a redesign eliminates that risk, the risk chart is updated to remove the risk from the chart. However, as with any document under document control, the earlier chart would be retained in the product file, with information indicating why the risk was removed. Figure 2 shows the use of a risk chart for the tracing of particular risk items throughout the life cycle.
In a risk chart, medical device reporting (MDR) event problem codes can be used at the intersection where the frequency and severity for a risk are indicated. The codes are listed in Appendix C of the Medwatch Medical Device Reporting Code Instructions Manual. The manual is available on-line at www.fda.gov/cdrh/mdr/373_appdxc.html. Use of these codes in the development process requires the input of clinical personnel into the risk management process. The input of clinical personnel is vital to developing an accurate risk analysis and should not impose any new burden (see Tables I and II).
|
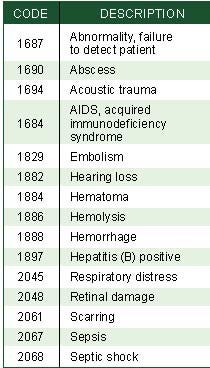
Table I. Patient-related codes for ear, nose, and throat products.(Click to enlarge). |
An event (e.g., a foreign material causing embolism by a product) could be represented as [1829, 1259]. When the event occurs, therefore, the complaint investigator could refer to these codes. The codes would link to a risk item on the device risk chart to easily locate the risk management information. Using a database, spreadsheet, or other tracking tool, a manufacturer could quickly determine whether the incident occurs with the predicted frequency and severity, as required on MDR form 3500A. If an event is inconsistent with the predictions, then a manufacturer knows which section of the risk analysis must be updated, as required by the standard.
The use of these codes in development simplifies the CAPA process analysis of the event and helps manufacturers to determine whether to take corrective actions. The information can also be input into a health hazard analysis used to determine whether a recall is necessary. If the event is occurring with the predicted severity and frequency, this information is documented and is also used as input to the verification of the effectiveness of the risk management process as required in Clause 3.3d of the ISO standard.
Other Postdevelopment Activities. During the life of a product, improvements and changes to the product are often made. This may include changes to reduce costs and to improve manufacturability. Any change to a product requires reconsideration of the risk analysis. When an inadequate design change process has been used, FDA has found situations in which changes to a product introduce new risks. A design change process must include a link back to the original risk analysis and indicate that persons with “knowledge and experience appropriate to the task” have reviewed the change.
|
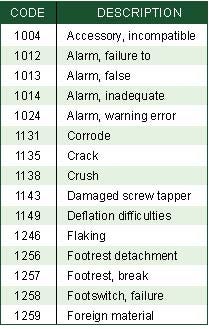
Table II. Device-related codes for ear, nose, and throat products (Click to enlarge). |
This review should be similar in nature to the original risk analysis. In fact, a company's design change procedure may refer back to the risk analysis procedure to accomplish this activity. It is critical that risk analysis activities be documented, even if the conclusion is that no change to the risk analysis will occur due to the design change.
It is crucial that changes to the manufacturing process are reviewed for changes to the risk analysis. Changes to the manufacturing process could expose patients and operators or others to new risks. Such changes could include the use of new manufacturing materials or cleaning processes.
Conclusion
To be most effective, postdevelopment risk management activities should be integrated with quality system activities. Most manufacturers already include risk analysis activities in their development processes. Adding postdevelopment activities to the quality system strengthens the system and improves a manufacturer's early warning of risk issues through reporting of events inconsistent with the initial risk analysis.
A manufacturer can also use post-development risk management activities to support the decision-making processes surrounding possible product recalls and the development of regulatory reporting documents.
Copyright ©2004 Medical Device & Diagnostic Industry
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
