Sign up for the QMED & MD+DI Daily newsletter.
A New Perspective on Medical Device Certification
In this proposed certification model, early collaboration can help manufacturers stay on track with standards requirements.
Mark Leimbeck
May 1, 2010
10 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "A New Perspective on Medical Device Certification")
The third edition of IEC 60601-1 will soon become de facto mandatory for certification of medical devices. This standard is unique, combining both product and process requirements into a single document. And by design, it specifically supports the innovation necessary for breakthrough technologies in the medical device industry. The standard accomplishes this by requiring manufacturers to utilize a product life cycle risk management process to understand and mitigate risks that are likely with any new technology or application. In short, a manufacturer must show that it has a risk management process in place and that all risks were identified and addressed such that they are acceptable according to the manufacturer’s policy on risk acceptability.
Formal assessments of a manufacturer’s risk management process will be a new activity for product certification bodies. ISO 14971 also requires a continuous life cycle approach—starting at design conception and following through to end of the life of the product. And because risk management requirements are woven throughout the fabric of IEC 60601-1 (there are approximately 115 references to “inspection of the risk management file”), certifiers and standards bodies such as the IECEE (IEC System for Conformity Testing and Certification of Electrotechnical Equipment and Components) may face a challenge in determining how best to perform assessments under these new requirements.
With product life cycle risk management becoming a new required element of certification, both the manufacturer and the certification body have a regulatory reason to explore new processes for delivering value to their customers. This situation also presents an opportunity to use advanced process design methodologies, such as lean, to create and optimize a certification method for this new, combined, product/process standard. ISO 14971 provides an internationally developed framework for risk management, which, by design, applies throughout the life cycle. This article discusses the details of the applicable requirements, and how lean process design techniques can be leveraged to create a new certification model. Such a model could be helpful in meeting various requirements, including IEC 60601.
Unique Complexity of the Device Industry
|
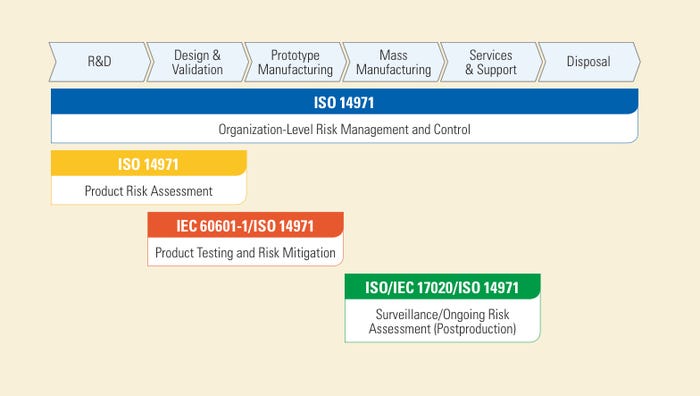
Figure 1. (Click to enlarge) Product life cycle certification model. |
The medical device industry faces numerous challenges in bringing a new product to market. Technological advances continue at an ever-increasing rate, bringing with them the opportunity for new methods to diagnose and treat patients. However, potential benefits in the diagnosis and treatment of patients must be balanced against harm that may occur. When all potential effects are not immediately understood, manufacturers of medical devices must maintain an ongoing process to identify, quantify, and mitigate risks. This process begins at the very earliest stages of product conceptualization and continues throughout the product life cycle, including the ultimate end of life for the product.
The latest generation of international consensus standards for medical device certification includes the following:
? ISO 14971, “Medical Devices—Application of Risk Management to Medical Devices.”
? IEC 60601-1, “Medical Electrical Equipment—General Requirements for Basic Safety and Essential Performance.”
? ISO 17020, “General Criteria for the Operation of Various Types of Bodies Performing Inspection.”
Many of the requirements in the standards are applicable at different times, and they represent discrete activities that must be completed from stage to stage. And although the discrete activities are completed at differing stages, they are nonetheless integrated and interdependent—forming a continuous process. ISO 14971 is clearly the main driver for this continuous safety assurance process. It states, “It cannot be emphasized too often that risk management does not stop when a medical device goes into production…With the postproduction information, the risk management process truly becomes an iterative closed-loop process.” Figure 1 illustrates the timing for the application of each of the standards noted above in the context of the product life cycle.
Note that ISO 14971 includes requirements for organization-level risk management and control. These requirements can be thought of as a sort of infrastructure that must be in place, not only before beginning a new product design, but throughout the entire life cycle of any product that may be produced. Additionally, this infrastructure supports not only individual products, but any range of products that may be produced by a manufacturer. The following elements are included:
? Management commitment (clauses 3.1, 3.2, 3.3, and 3.4).
? Risk management system support (clauses 3.5, 3.6, and 8).
? Risk management process and control (clauses 4, 5, 6, and 7).
? Process and product monitoring (clauses 3.3 and 9).
Additionally, ISO 14971 includes product risk assessment requirements for manufacturers to apply the above infrastructure in identifying, analyzing, evaluating, and mitigating the risks associated with a specific product. These activities typically first occur during the manufacturer’s research and development and design and validation stages, but per the ongoing process required by the infrastructure, they are continuously reassessed based on field experience. The specific requirements include:
? Device-specific risk management plan (clause 3.5).
? Risk management traceability matrix (clause 3.6).
? Risk analysis (clause 4).
? Risk evaluation (clause 5).
? Risk controls (clause 6).
? Overall risk evaluation (clause 7).
? Risk management report (clause 8).
As the product moves through the design and validation and prototype manufacturing stages, IEC 60601-1 and ISO 14971 are jointly applied in product testing and risk mitigation. In this joint application of requirements, certain decisions that are dependent on the risk management process must be made, such as the following:
? Applicability of specific requirements.
? Options for risk control.
? Modification of specific tests.
? Selection of particular tests.
? Changes to pass-fail criteria (subject to clause 4.5).
Once into the mass manufacturing and service and support product life cycle stage, the surveillance and ongoing risk assessment requirements from ISO 17020 and ISO 14971 apply. ISO 17020 is a standard that provides requirements for certifiers performing inspections of products, processes, and work procedures. The purpose of the inspections is to determine the ongoing conformity of products, processes, and work procedures with requirements. The results of these activities are subsequently reported to clients and, when required, to supervisory authorities (regulators). This standard notes that inspections of a product or facility may concern all stages during the lifetime of these items, including the design stage. This is an important consideration because, as previously noted, ISO 14971 is applicable to products and organizations throughout their life cycle.
Certification Process
In sum, the standards have introduced new requirements for the manufacturer’s process steps that must now be included as part of the assessment performed by conformity assessment bodies. (In this context, an assessment is the method by which conformance with a given set of requirements is established for a process or device. Without being prescriptive, the method could be a document review of objective evidence, an on-site audit, or other means.) Ideally, a new certification model would integrate product and process requirements, appropriately time the discrete activities associated with each of the OEM’s process steps, and continuously apply all steps well after the initial certification and product launch. To that end, lean process design offers some help in developing adjustments to the certification process.
Applying Lean Principles to Product Certification
As a medical device moves through the life cycle and approaches product launch, many key decisions are made by the manufacturer regarding not only basic form and functioning, but also specific materials, subassemblies, manufacturing tooling and dies, manuals, and other facets. Once these decisions are made, any revision to a product can increase costs exponentially due to a cascading effect for decisions that are dependent on earlier decisions. Needless to say, significant opportunity costs may also be incurred when and if launch dates are compromised. These factors are a key motivator for one of the teachings of lean: to ensure 100% quality throughout the process.
Other teachings of lean are applicable to the process as well—for example, the concept of continuous flow. ISO 14971 almost seems to suggest this approach as a requirement with its emphasis on developing a true “iterative closed-loop process.” Perhaps most important is the lean principle of designing the process around value-adding activities. This requires an understanding of which activities are truly value-adding from the final customer’s perspective, i.e., the patient or end-user. It is difficult to imagine what could be more value-adding than safety from the perspective of a patient or user, thus the importance of considering risk management throughout the life cycle.
An Adjusted Model
In applying these principles to medical device certification, the discrete activities previously described and depicted in Figure 1 would include, as near as possible, real-time validation of conformance with applicable requirements. Medical device complexity, as well as the new regulatory environment, would suggest ongoing vigilance as the most prudent course of action when it comes to product realization.
In practice, therefore, adjustments to the certification model would include some level of integration of the manufacturer and certifier tasks, performed as early in the product development cycle as possible. This model would potentially include the following elements.
Early Dialogue. One critical aspect is an early and planned continuing engagement between the manufacturer and the certifier. This communication is intended to reduce uncertainty regarding information needed by a certifier to begin an investigation (ensure 100% quality), inherent delays in acquiring and submitting the information (continuous flow), and multiple non-value-adding administrative start-up activities if and when design iterations are resubmitted (continuous flow).
Concurrent Certification Reviews of Design Options. As a design staff develops options and alternative product configurations, they may interpret requirements differently from certifiers, often leading to rework. By moving the certification review to be concurrent with interpretation and application of requirements by the designer, certification engineers can clarify and align interpretations, thus eliminating a costly source of rework and delays (ensure 100% quality, continuous flow, design around value-adding activities).
Simultaneous Design and Test Program Development. As the design progresses, the integration of tasks with certifier assessments allows real-time feedback about design options as well as trade-off analyses regarding test programs, development timelines, and other considerations (ensure 100% quality, continuous flow, design around value-adding activities).
Integrated Development and Certification Testing. Development testing results may qualify for certification testing (with ISO 17025–based controls). This eliminates redundant testing and additional waiting time (continuous flow).
Final Review Checklist. Because the bulk of value-added engineering work is performed early in the product life cycle, much of the final review prior to issuing certification is validating the compliance of testing results; that is, earlier assessment activities will have already validated compliance with constructional and risk management requirements (ensure 100% quality, continuous flow, design around value-adding activities). The difference in final review is a key enabler for meeting schedules, since the most costly and significant time delays associated with certification are issues identified at the final step in the process prior to certification.
ISO 14971 Certification. As already noted, ISO 14971 is a continuous life cycle approach to product safety. After initial certification, the manufacturer continually seeks to identify new information that will lead to enhanced product safety, and when found, closes the loop by feeding this information back into the continuous risk management process for action. Ongoing assessments of this process validate the manufacturer’s continued conformance with these requirements and serve as evidence to regulators of compliance (ensure 100% quality, continuous flow, design around value-adding activities).
Following this model may significantly reduce compliance issues and overall product development cycle time. Both the manufacturer and the certifier benefit from enhanced communication and placing technical expertise where it adds the most value in the overall process flow. Additionally, such a model would enable demonstrated compliance with internationally recognized consensus standards.
Conclusion
With the adoption of the third edition of IEC 60601-1, many devices will need to be recertified, some products will need to be redesigned, and entirely new products continue to be developed. As the watershed event of the first standard requiring a life cycle risk management process continues to approach, a certification model that additionally integrates and applies lean process improvement would be beneficial to manufacturers.
Mark Leimbeck is program manager, health sciences, at Underwriters Laboratories Inc. (Northbrook, IL).
About the Author(s)
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
