Sign up for the QMED & MD+DI Daily newsletter.
Serious Adverse Events Triple in Three Years: Is Risk or Reporting to Blame?
April 25, 2012
6 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Serious Adverse Events Triple in Three Years: Is Risk or Reporting to Blame?")
By Nora Iluri, founder and CEO, Clarimed and DeviceMatters
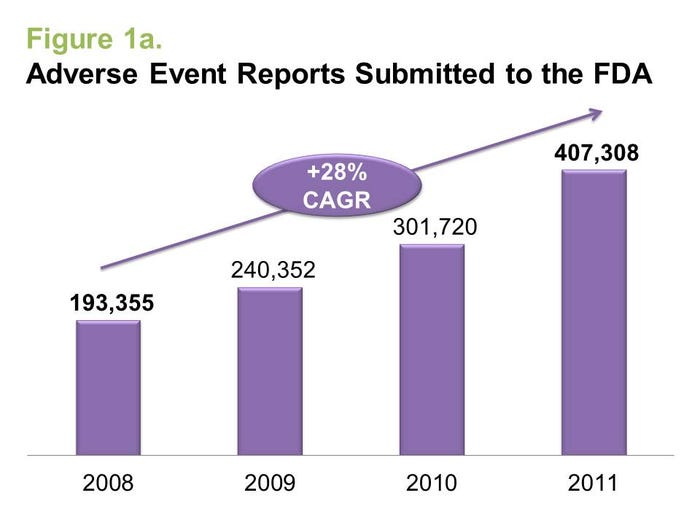
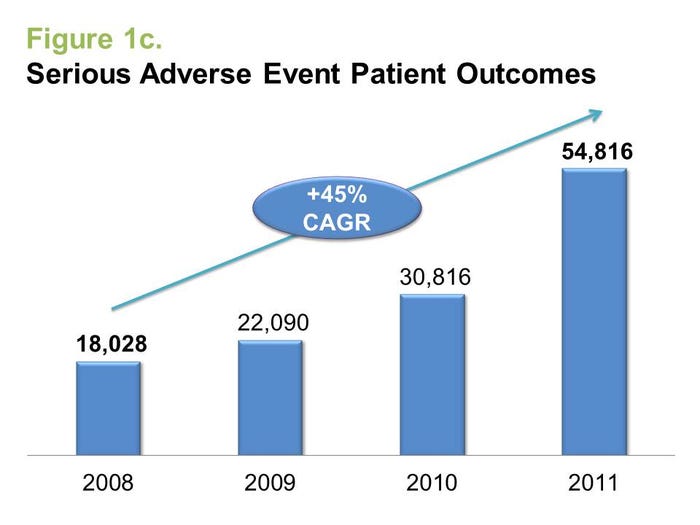
Doubling during the past three years, more than 400,000 medical device adverse event reports were submitted to FDA in 2011, according to healthcare rating agency DeviceMatters. Perhaps even more shocking, however, is the 45% annual growth rate and more than 50,000 reported serious patient outcomes--defined as hospitalization or worse--associated with medical device malfunctions or defects in 2011. But is this rapid rise in recent adverse events indicative of riskier medical devices entering the market, or is the industry just becoming more diligent about reporting them?
Key Drivers of Adverse Event Growth
Adverse event reports are filed when a patient has been injured or could have been injured while using a particular medical device. While these reports do not necessarily prove that a medical device caused an injury, they do provide a means of assessing the risk involved in using the product.
|
|
|
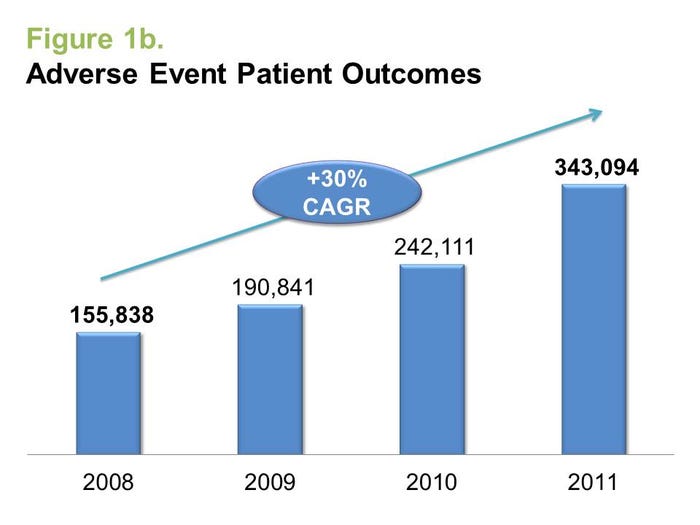
On occasion, multiple reports can be filed for the same adverse event. Some adverse events, however, will not involve a patient report. As such, patient outcomes are considered to be a more-accurate measure of the real patient impact of adverse events. In reviewing this data, DeviceMatters found that the top five device categories with the most adverse events (AEs) in 2011 were:
Needles, Syringes, and Infusion Supplies: 53,500 AEs; 3400 serious AEs
Pacemaker and Defibrillator Pulse Generators: 35,100 AEs; 16,600 serious AEs
Sugar (Saccharide) Test Systems: 30,300 AEs; 5700 serious AEs
Pacemaker and Defibrillator Electrodes: 23,500 AEs; 9700 serious AEs
Hip Implants: 25,200 AEs; 2300 serious AEs
These statistics might, at first glance, appear alarming. However, there are four key drivers for the growth in adverse event reports that may justify the jump and even reflect positively on the industry.
Driver 1: Improved Reporting Compliance
Many experts believe that adverse events are vastly underreported and that much of the growth observed in recent years is simply indicative of improved reporting. Thus, the case may be that the number of people experiencing adverse events is not increasing, but rather that these events are more likely to be reported when they do occur. If the increase in adverse events is purely attributed to improved reporting, then consumers have nothing to worry about.
Driver 2: More Devices in Use Means More Adverse Events
Medical device prevalence has also grown significantly faster than the healthy 6 to 9% CAGR of the medical device market because products such as pacemakers, defibrillators, and hip implants are expected to have a lifetime ranging from 10 to 20 or more years. Unfortunately, we do not centrally track devices in use--not even for high-risk devices. Therefore, we cannot calculate the normalized adverse event rate for each device category.
Driver 3: Increased Complexity Leads to Errors
Medical devices are becoming increasingly complex, and complexity spawns errors. This notion is particularly evident in the increased use of electronics and software in recent years as well as in the associated threats, such as malware, that are growing exponentially each year.
Driver 4: Higher-Risk Patient Groups and Procedures Result in Increased Adverse Events
Finally, as medical device manufacturers continue to innovate, they often target patients that were previously considered too frail or unsuitable for other medical devices or procedures. Percutaneous heart valves, for example, were introduced specifically for use in some of the most vulnerable patients. If the patient is very sick or the procedure is especially risky, adverse events are more likely to occur. Nevertheless, the benefits might still be worth the risk for those patients.
Regulatory Red Flags
Taking these four factors into account, it is difficult to determine whether overall medical device safety across the industry is actually improving or declining. Nevertheless, the data provides us with ways to evaluate the impact of some of these drivers and gain a better understanding of which categories deserve more or less regulatory attention.
It is interesting to note, for example, that the majority of adverse events hail from a very small number of product groups. We at DeviceMatters have classified medical devices into almost 600 product groups; five of these product groups (listed previously) were responsible for 50% of all adverse event patient outcomes in 2011.
In terms of the growth seen in adverse event outcomes from 2010 to 2011, the same five groups are responsible for 80% of the growth. But if we exclude these five product groups, adverse events for the rest of the medical device industry are pretty much in line with industry growth.
Upon reviewing this data, the question becomes: Are adverse event reports a self-fulfilling prophecy of media and regulatory attention, or can they be used as a predictive measure by which the report numbers tend to skyrocket once issues are reported?
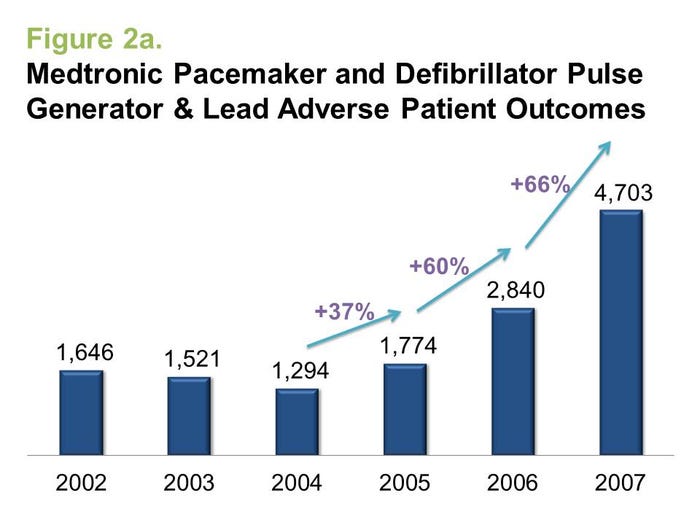
To determine the answer, we need to compare when adverse events began to rise with the timing of a media and regulatory attention-grabbing event such as a major recall. As one example, we examined the adverse event reports filed for Medtronic's pacemakers and defibrillators prior to the recall of the Fidelis lead in 2007.
|
|
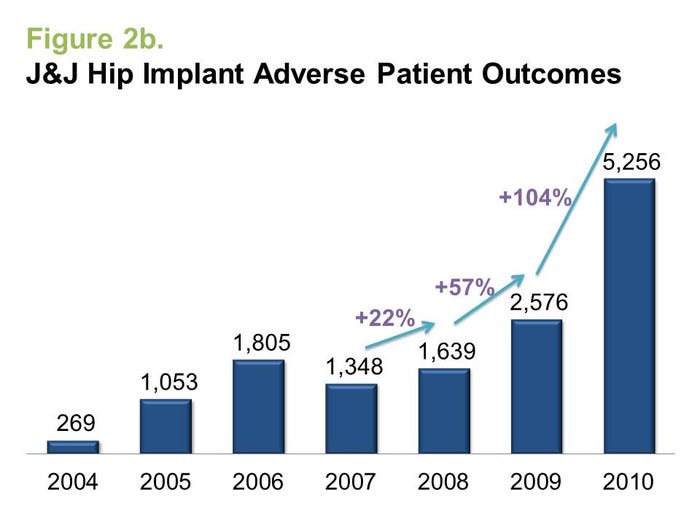
As seen in Figure 2a, the growth rate of adverse events was, in fact, quite significant prior to the recall of the Fidelis lead in 2007, although the recall did accelerate reporting beyond previously seen rates. Likewise, adverse events for DePuy's metal-on-metal hip implants began rising in 2007, a full three years prior to the ASR recalls (Figure 2b).
Exploiting Adverse Event Data
While adverse event analytics may not be the silver bullet, analyzing adverse event data in a new light can definitely help provide new insights and early warnings for areas that should be further investigated. Reporting and analytics are starting to be mature enough to serve as an early-warning system to signal potential problems, even though understanding underlying drivers and causes will require additional research in each case. Given that these data are the only source of such fact-based statistics, they should be harnessed, used, and improved upon.
And industry experts and the FDA can all agree upon that final point. As FDA noted in an October 2011 white paper titled "Understanding Barriers to Medical Device Quality:" "Moving toward greater visibility into device quality and properly aligning FDA's regulatory approach will be important to catalyzing industry movement towards improved device quality."
Nora Iluri is founder and CEO of Clarimed and DeviceMatters, which focus on medical device safety and approvals with the intention of fostering innovation and improvements in quality. Clarimed has partnered with Qmed to offer users exclusive access to download reports by product code, device category, or by manufacturer through the DeviceMatters Web site. Users can find and download free reports on any device category or manufacturer. Full, detailed reports are also available for a fee. DeviceMatters is currently the only Web site that provides adverse event reporting of medical devices based on unbiased and independent FDA data, among other sources.
You May Also Like

.png?width=300&auto=webp&quality=80&disable=upscale)
