






Sign up for the QMED & MD+DI Daily newsletter.
A Primer for IEC 60601-1
The process of "designing in" safety is a must to ensure that electrical medical products succeed in meeting both regulatory requirements and market prospects. Projects are often delayed due to underestimating the product safety requirements in the initial design phase.
+1
18 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "A Primer for IEC 60601-1")
|
Leonard Eisner |
|
Robert M. Brown |
|
Dan Modi |
This oversight can be costly to the medical manufacturer in terms of product design expenses, compliance testing and certification turnaround time, and the device approval process. Medical products must go through compliance testing and device approval before they can be marketed.
The IEC 60601-1 standard, Medical Electrical Equipment—Part 1: General Requirements for Safety, is the cornerstone document addressing many of the risks associated with electrical medical equipment. Ensuring that a device complies with IEC 60601 can be a complex, multifaceted task. This article presents an overview of the current requirements to assist design engineers, R&D engineers, compliance engineers, and regulatory and quality affairs personnel in meeting this challenge.
What products fall under this standard? Electromedical products are defined in IEC 60601-1 Subclause 2.2.15 as "equipment, provided with not more than one connection to a particular supply mains and intended to diagnose, treat, or monitor the patient under medical supervision and which makes physical or electrical contact with the patient and/or transfers energy to or from the patient and/or detects such energy transfer to or from the patient." Examples of products fitting this definition include battery-operated thermometers, MRI and gamma imaging systems, endoscopic cameras, infusion pumps, and many others. Accessories used with such equipment can also fall under this standard.
Technical Committee (TC) 62 of the International Electrotechnical Commission (IEC) publishes the international IEC 60601-1 standard. The scope of TC 62 is electrical equipment in medical practice. The facilitator in the United States for IEC 60601-1 is the American National Standards Institute (ANSI; www.ansi.org). IEC 60601-1 is currently at the second edition, published in 1988, and has undergone two amendments; the first amendment in 1991, and the second in 1995. The first edition of the standard was published in 1970.
|
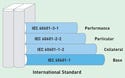
Figure 1. Structure of the IEC 60601 international standard. |
The IEC 60601 Standard
The IEC 60601 standards series consists of four distinct parts (see Figure 1). The IEC 60601-1 base standard is the core of the series and a part of the 60601–1 grouping (base and collateral). The 60601–2 grouping includes particular device-specific standards, and the 60601–3 grouping includes performance and device-specific standards.
Base Standard. IEC 60601-1 covers all the general requirements for electrical medical (or electromedical) products.
Collateral Standards. Standards numbered IEC 60601-1-x contain horizontal issues that may deal with many different types of medical devices. IEC 60601-1-2 is an example of a collateral standard, and it encompasses electromagnetic compatibility (EMC) issues of electrical medical devices. A standard on the horizon in this category is IEC 60601-1-6, which deals with human factors (usability) issues. Members of the Association for the Advancement of Medical Instrumentation (AAMI; www.aami.org) can obtain drafts of the IEC standards to prepare for changes that may impact the manufacturer's product design.
Particular Standards. Standards numbered IEC 60601-2-x lay out requirements for a specific type of medical device. IEC 60601-2-2 is the particular standard for high-frequency surgical devices. Particular standards can amend, modify, and/or supersede part of the requirements specified in IEC 60601-1.
Performance Standards. Standards numbered IEC 60601-3-x lay out performance requirements for specific types of devices. IEC 60601-3-1, for example, contains "essential requirements for the performance of transcutaneous oxygen and carbon dioxide partial pressure monitoring equipment."
|
Figure 2. IEC 60601 and national standards. |
IEC 60601 and National Standards
As illustrated in Figure 2, the base standard IEC 60601-1 has been adopted as a national standard in most major countries. The standard, either as national standard (such as JIS T0601-1 in Japan) or as the base IEC 60601-1 itself (e.g., in Brazil), is accepted in nearly all markets for supporting regulatory registrations or approvals (see Table I). It should be noted that in the United States, UL 2601-1 has been changed to UL 60601-1, 1st edition, titled Medical Electrical Equipment, Part 1: General Requirements for Safety, published April 25, 2003. There are no changes to the requirements from UL 2601-1. There is a change in the formatting of the standard. All the U.S. deviations as well as amendments 1 and 2 of IEC 60601-1 are combined within the body of the standard.
COUNTRY | IEC 60601-1 ADOPTED AS: |
United States | ANSI/ UL 2601-1 (U.S. national deviations) |
Canada | CAN/CSA C22.2 No. 601.1 |
European Union | EN 60601-1 (identical to IEC 60601-1); in UK, BS EN 60601-1 |
Japan | JIS T0601-1 (Japanese national deviations) |
Astralia/New Zealand | AS/NZ 3200.1 (Australian and New Zealand national deviations) |
Table I. IEC 60601-1 national standards |
Other Medical Standards
Although there are other national medical standards, IEC 60601-1 is the governing standard for electrical medical products. In the United States and Canada, UL 544, UL 187, and CAN/CSA C22.2 No. 125 and No. 114 will all be withdrawn January 1, 2005.
Manufacturers who currently certify their medical products to UL 544, UL 187, CAN/CSA C22.2 No. 125, or No. 114 will need to remove the certification mark as of January 1, 2005. Manufacturers may need to recertify to UL 2601-1 (United States) or CAN/CSA C22.2 No. 601.1, respectively, depending on the product design.
Now is a prudent time to review the product design and the requirements of IEC 60601 (plus national deviations) to determine if additional product compliance testing and recertification are necessary for the January 1, 2005, transition.
Global Regulatory Importance of IEC 60601-1
Compliance with IEC 60601-1 and/or a national standard does not equal medical device approval. Compliance is step one, approval step two, and marketing step three. The Global Harmonization Task Force (GHTF; www.ghtf.org), established by the United States, Canada, Australia, Japan, and the European Union, gives credence to using the IEC 60601-1 standard as the model for compliance of electrical medical devices.
United States. FDA is the approval agency, and its Center for Devices and Radiological Health (CDRH) oversees the regulation of electrical medical devices. FDA recognizes IEC 60601-1 as a consensus standard with any amendments, and with specific national alterations, such as ANSI/UL 2601-1. More information regarding CDRH's consensus standards can be found at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm.
The agency has stated that conformance with recognized consensus standards like IEC 60601-1 can provide a reasonable assurance of safety for many applicable aspects of a medical device and has direct bearing on safety determinations made during FDA's premarket application reviews. The premarket application process may include: premarket notification (510(k)), investigational device exemption (IDE) application, premarket approval (PMA) application, humanitarian device exemption (HDE) application, or product development protocol (PDP).
With the use of a consensus standard, a submission can contain a declaration of conformity to that standard and eliminate the need to submit the bulk of test data for those aspects of the device addressed by said consensus standard. As a consensus standard, IEC 60601-1 also allows manufacturers of electromedical products to use the abbreviated 510(k) paradigm where appropriate. Information on the 510(k) paradigm can be found at www.fda.gov/cdrh/ode/parad510.html.
European Union. The European Union (EU) includes 15 member states. They are Austria, Belgium, Denmark, Finland, France, Germany, Greece, Ireland, Italy, Luxembourg, the Netherlands, Portugal, Spain, Sweden, and the United Kingdom. The European Economic Council (EEC) publishes the Medical Devices Directives (MDD; 93/42/EEC; http://europa.eu.int/comm/enterprise/medical_devices/index.htm), which declare EN 60601-1 (identical to IEC 60601-1) a harmonized standard. For products complying with EN 60601-1, this declaration gives a "presumption of conformity" to a large majority of the essential requirements of the directives. Meeting the essential requirements is a major step toward receiving a CE mark for a device.
The MDD allow self-certification to the standard under a recognized quality system such as ISO 9001:1994 with ISO 13485:1996 (previously EN 46001). The evaluation is subject to review by a notified body as part of the technical file or design dossier review. A notified body may be required for testing of the manufacturer's medical device(s), certification of the manufacturer's quality system, or review of the technical file or design dossier for the medical device based on the classification of the product.
Each notified body is given notice by publication in the Official Journal of the European Community (OJEC) of the specific areas of competence that the competent authority has verified said notified body is able to provide. It is important to note that some notified bodies are only allowed to perform specific testing and/or quality system registration. Some notified bodies are allowed to work on all product classes, perform product testing, and perform quality system registration. Prior to selecting a notified body, manufacturers should confirm what they have been notified for by checking with the OJEC.
Canada. The Therapeutic Product Directorate (TPD; www.hc-sc.gc.ca/hpfd-dgpsa/tpd-dpt/index_e.html) of Health Canada is the Canadian approval agency and recognizes CAN/CSA C22.2 No. 601.1 as the regulatory compliance standard for electrical medical devices. Canada's approval system is similar to that of the EU. The major differences are the registration process, the classification system, the postmarket surveillance method, and the quality system standards used.
Four classes of medical electrical devices are recognized, and registration is required for Class II, III, and IV devices (Class IV being the highest-risk class). Importers, distributors, and manufacturers of Class I devices must get an establishment license.
The quality system requirements are ISO 13485 for Class III and IV devices, and ISO 13488 for Class II devices. A third-party auditing firm accredited by the TPD conducts the certification of the quality system. Class I devices do not require a quality system.
Japan. The Ministry of Health, Labor and Welfare (MHLW; www.mhlw.go.jp/english) is the regulatory agency in Japan. JIS T0601-1 is recognized as the compliance standard to support registration of electromedical products. The Japanese Association for the Advancement of Medical Equipment (JAAME) is a designated agency appointed by MHLW under the Pharmaceutical Affairs Law. Evaluation data from JAAME or a foreign equivalent organization (such as Underwriters Laboratories or TUV Product Service) based on the JIS T0601-1 standard are accepted in the medical device approval process. Medical devices requiring clinical studies for approval will also have data collected on the product for three years after the product is released, per the MHLW postmarket safety assurance program.
Australia. The Therapeutic Goods Administration (TGA; www.health.gov.au/tga), a part of the Federal Department of Health and Ageing, is the regulatory agency in Australia. Medical devices go through a premarket assessment and are assigned an AUST R number in the Australian Register of Therapeutic Goods, or ARTG. The ARTG is the computer database for therapeutic products approved for supply in or export from Australia.
Manufacturers of therapeutic goods must be licensed and their processes must be compliant with good manufacturing practices, or GMPs. Postmarket surveillance of products includes investigation of failures, laboratory testing, and monitoring for compliance with legislation.
The use of standards to support regulatory approval and registration is voluntary, but the use of IEC 60601-1 or AS/NZ 3200.1 to support the electrical safety portion of an application has been acceptable in Australia for some years. The mutual recognition agreement (MRA) between Australia and the EU is finalized. This allows the TGA to permit some EU-notified bodies to assess products and companies in light of the Australian regulations. The Australia EU MRA can be found at www.ecdel.org.au/eu_and_australia/agreements_mra.htm and http://www.tga.gov.au.
Other Countries. The regulatory environment is growing in Pan-Asian and South American countries. Countries are adopting regulations for medical devices, or increasing the enforcement of regulations already on the books. Most of the countries are following the model of the GHTF. The GHTF model is similar to that of the EU, and gives importance to the recognized (international) standards for showing compliance to the essential principles of safety and performance/efficacy. Most of the countries that have established regulations (and enforce them), such as Korea, Brazil, and Argentina, recognize the IEC 60601-1 standard for showing compliance of electromedical products to the general safety requirements of the regulation.
Structure of the Base Standard
The structure of the base IEC 60601-1 standard is hazards-specific. It provides requirements for evaluating the common hazards associated with electromedical products. Its scope is to protect both patients and users by reducing the likelihood of the following hazards.
Electrical Shock Hazards. Reduce exposure (access to the user or the patient) to voltages exceeding 25 V ac or 60 V dc, energy hazards, and/or excessive allowable leakage currents. Provide for separation of circuits, proper grounding, and meeting the appropriate dielectric tests. Refer to section 3 of IEC 60601-1.
Mechanical Hazards. Reduce exposure to moving parts, pinching, crushing, overtilt, expelled parts, dropping, supports breaking, and others. Refer to section 4 of IEC 60601-1.
Radiation Hazards. Reduce the risk of x-radiation exceeding 0.5 mrd in a one-hour period at a distance of 5 cm from accessible surfaces outside the treatment zone. Refer to section 5 of IEC 60601-1. For EMC, refer to the collateral standard, IEC 60601-1-2. IEC 60601-1-2, second edition, introduces the concept of essential performance which is being incorporated into the draft third edition of IEC 60601-1. Refer to the basic concept section.
Ignition Hazards of Flammable Anesthetics. Reduce the exposure of flammable anesthetics to static discharge, corona discharge, high-energy circuits, and restricted ventilation, among others. Refer to section 6 of IEC 60601-1. Note that flammable anesthetics are seldom used today.
Fire and Other Hazards. Reduce the exposure to excessive temperatures, liquid spillage, pressure vessels, human errors, and other such hazards. For biological hazards (biocompatibility), refer to the international standard ISO 10993-1. For detailed requirements on human errors, refer to the appropriate IEC 60601-2-xx standard specific to the device under test. Refer to section 7 of IEC 60601-1.
Excessive (Energy) Output Hazard. Reduce exposure caused by inaccuracy of operating data or the accidental high setting of output. For detailed requirements on excessive output, refer to the IEC 60601-2-xx standard specific to the device under test. Refer to section 8 of IEC 60601-1.
Sections 1 and 2 of IEC 60601-1 address the general requirements for tests (such as definitions and classification) and environmental conditions (including temperature, humidity, supply voltage, and others). Section 9 identifies abnormal and fault conditions which must be evaluated. Some foreseeable failure conditions include blocked vents, locked fan rotor, and short and overload of isolation transformers. Section 10 addresses the general construction requirements for enclosure, components, and grounding (or earthing) that are not included in the other sections, yet support the requirements of those sections.
Basic Concept
IEC 60601-1 requires that two levels of protection be employed in various areas of the product to meet the requirements of the standard. If one level of protection fails, the product would then still have another level of protection to contain any electrical shock hazards and shield patients and operators from harm.
IEC 60601-1 permits three building blocks to be used in various combinations to meet the "two levels of protection" requirement. These building blocks are insulation, protective earthing, and protective impedance. For example, a protective earth (one level of protection) used in combination with basic insulation (one level of protection) provides the two levels of protection that are required. Alternatively, a product's plastic enclosure that has reinforced insulation (considered two levels of protection) between the outside of the enclosure and its circuits again achieves two levels of protection.
IEC 60601-1 is based on the same concept as risk management. That is, to assess and control risks in the product design, manufacture, and intended use. IEC 60601-1 uses one or more of the following risk-control measures: it forces inherent safety by design, it imposes protective measures in the medical device or its manufacturing process, or it requires instructions and/or labeling information for safety.
The draft third edition of IEC 60601-1 cites the international risk management standard ISO 14971. The third edition of IEC 60601-1 is at the committee draft for vote (CDV) level of the standards development process. The first committee draft vote (CDV-1) failed to attract a positive vote. It is hoped that a second CDV will be voted on before the end of 2003, after the September 22–October 2, meeting of Subcommittee 62A in Frankfurt, Germany.
The third edition broadens the concepts in the second edition (of basic safety) and adds requirements of essential performance to the standard. These are performance characteristics necessary to maintain residual risk (risk after protective measures are taken) within acceptable limits.
The third edition will have two types of requirements. These include requirements needing evaluation based on tests or document review—and not requiring a risk analysis, and requirements where evaluation requires the manufacturer to conduct risk analysis.
Classification
IEC 60601-1 specifies requirements based on the product classification. The classification of the medical product must be determined first in order to proceed with the class-specific requirements of the standard. Product classification is based on different criteria in terms of safety and intended use. Classification criteria include the following.
Protection against Electrical Shock. For devices powered by an external source, the product may be classified as Class I or II. Class I is a product that is provided with a reliable protective earth (PE), such as a complete metal enclosure, that is protectively tied to the ground pin of the three-pronged power plug. Construction is such that accessible metal parts cannot become live in the event of a single fault. Class II is a product without a PE and where double or reinforced insulation is relied upon to provide protection against electric shock. For example, a product has an external brick power supply that provides double insulation. The Class II symbol is a double-walled square, indicating the product's double insulation.
|
Figure 3. Classification of degrees of protection against electric shock. |
Degree of Protection (Applied Part) against Electric Shock. This product classification deals with the definition of applied parts—those parts or circuits that deliberately come in physical contact with the patient. The classification applies to each applied part. They are classified either as type B, BF or CF, depending on the degree of protection they offer against electric shock (see Figure 3).
Degree of Protection Against Ingress of Liquids. This classification deals with device construction to protect it from the entry of a liquid. It is identified by IP code as specified by IEC 60529. In most cases, other than foot switches, it is up to the manufacturer to determine the IP rating and pass the appropriate level tests (see Figure 4).
|
Figure 4. Classification of degrees of protection against liquid ingress. |
Use with Flammable Anesthetics. There are three classifications for products that depend on their compatibility with flammable anesthetics (see Figure 5).
There are seven classifications based on the installation or use of the product. They are handheld, mobile, portable, transportable, stationary, permanently installed, and fixed equipment. Although some of these words seem identical in terminology, they have distinct definitions within the standard.
|
Figure 5. Classification of products by compatibility with flammable anesthetics. |
IEC 60601-1 also defines five modes of operation. These include continuous, short-time, intermittent, continuous operation with short-time loading, and continuous operation with intermittent loading. The most common classification of operation is continuous. The other four modes of operation limit the range in which the product is utilized, and the product is certified with those limits of use.
Conclusion
In this world of numerous regulatory requirements for medical products, manufacturers need to be able to get their products to market quickly, efficiently, and with the lowest expense in order to make a profit.
Planning a project before product testing through a third-party test house (such as UL, TUV, BSI, or CSA) is a critical factor in ensuring project success.
Incorporating IEC 60601-1 and the national deviations is step one in meeting global regulations for electromedical equipment. Understand the standard requirements well, design and evaluate the product to the standard, then complete third-party testing. An in-house certification engineer, a consultant that specializes in electromedical products, or some nonconsulting help from a test house can help solve pretesting problems. A test house cannot provide the manufacturer with consulting services because this is a conflict of interest. Step two, product approval, is accomplished with less difficulty when compliance with the global standard is complete.
Copyright ©2003 Medical Device & Diagnostic Industry
You May Also Like


