Sign up for the QMED & MD+DI Daily newsletter.
Integrating Risk Management with Design Control
Medical Device & Diagnostic Industry Magazine MDDI Article Index An MD&DI October 1998 Column RISK MANAGEMENT Making a risk management program part of a company's design control procedures satisfies FDA and improves device safety.
October 1, 1998
10 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Integrating Risk Management with Design Control")
A study of all voluntary medical device recalls in fiscal years 1983–1989 observes that "44% were attributed to errors or deficiencies that were designed into particular devices and may have been prevented by adequate design controls."1 Effective risk management carried out as part of a design control program can help reduce the problems that lead to recalls.
DEFINING RISK MANAGEMENT
Risk management is the systematic application of management policies, procedures, and practices to identifying, analyzing, controlling, and monitoring risk.2 The four goals of risk management are to identify any design or process inadequacies that could reduce safety or performance, eliminate these inadequacies or minimize their effect, evaluate the controls and remedies used to do so, and document the procedures.
The differences between risks and hazards have been defined by ISO/DIS 14971.3 A hazard is a potential source of harm—either physical injury or damage to health or property. Risk is the probable rate of occurrence of a hazard causing harm and the severity of the harm. Safety is freedom from an unacceptable risk of harm.
Addressing safety-related risks occurs in three stages. Risk analysis is the identification of hazards and the estimation of risks. Risk evaluation is the appraisal of identified risks and associated risk reduction alternatives. Risk reduction and control is the making and monitoring of risk reduction decisions over the product's life cycle. The cumulative nature of these activities is illustrated in Figure 1.3
 Figure 1. Relationship between activities to address risk. Taken from ISO/DIS 14971 and reproduced with the permission of the International Organization for Standardization (ISO).
Figure 1. Relationship between activities to address risk. Taken from ISO/DIS 14971 and reproduced with the permission of the International Organization for Standardization (ISO).
Several types of analyses can be used to integrate risk assessment activities into the product life cycle.
A system-level assessment analyzes the overall system and its major subsystems during the early concept and design stage. It looks for potential failure modes caused by system deficiencies and subsystem interactions.
A design assessment focuses on the product's design before it is released to manufacturing. It addresses design intent by reviewing design requirements and design alternatives.
A process assessment analyzes manufacturing and assembly processes. It examines the effects of tools, processes, machines, and assembly and manufacturing procedures on the design.
A service assessment focuses on failure modes caused by system or process deficiencies in a service function before the product reaches the customer.
These assessments fit into the product life cycle as illustrated in Figure 2.
 Figure 2. Risk assessments in the product life cycle.
Figure 2. Risk assessments in the product life cycle.
INTEGRATING RISK MANAGEMENT
FDA's quality system regulation for medical devices states that "design validation shall include...risk analysis, where appropriate" (820.30(g)). Where appropriate is defined as "appropriate unless the manufacturer can document justification otherwise."4 From a practical standpoint, risk analysis is always appropriate for establishing the rationale behind decisions on device safety.
For example, the manufacturer of a conductive gel for an electrocardiogram (ECG) system argued that its product had such an inherently low risk that a risk analysis was not necessary. If the gel failed to perform, the ECG monitor would not pick up a signal, which the operator would immediately notice. No harm would come to the patient. After a very brief preliminary hazard analysis, however, the manufacturer discovered that if the gel was not biocompatible, it could cause irritation (i.e., harm) to a patient. Thus, the risk analysis of an apparently harmless gel identified a potential source of harm worthy of assessment.

Figure 3. Integration of risk assessment with design control activities.5
Although risk analysis is addressed in the design validation section of the regulation, FDA expects risk analysis activities to be integrated with design control.2 Figure 3 shows how risk assessment can be integrated with the following design control activities:
Project Planning. A risk management plan should describe the strategic approach to identifying and controlling risk in the product development life cycle. This plan could be a part of the product development or project management plan.
Design Input. Existing safety standards and safety requirements identified in risk assessments are key design inputs.
Design Output. Risk reduction measures introduced into product design are essential design outputs.
Design Verification. Design verification should affirm that all safety requirements are covered by the risk reduction measures in the design.
Design Validation. Design validation should demonstrate that all safety requirements can be consistently met with respect to intended use and user or patient needs.
Design Transfer. Each scheduled design review should address risk assessment activities, as needed, to ensure that recommended actions are assigned and monitored prior to design transfer.
The product life cycle does not end with design transfer. Risk management activities also should be integrated with production and distribution. Component and process controls should ensure that safety requirements are met. Nonconforming products should be evaluated to make certain all risks were accounted for. Corrective or preventive action, especially in reaction to complaints, must be documented and evaluated for effectiveness.
IMPLEMENTING RISK MANAGEMENT
The FDA and ISO guidelines do not prescribe how to implement risk management. Manufacturers have the freedom to determine the policies and procedures that are best for them.
Risk management should be part of a firm's strategic planning. Companies must define their risk management policies and goals. For sufficiently large projects, a risk management plan should be created to guide risk assessment activities throughout product development. New procedures may be needed to support risk management; for example, scheduled reviews (i.e., requirements, design, test, product release) should include critical reviews of specific risk assessment activities.
Risk assessment activities need to be carefully planned and coordinated. This planning can be facilitated by establishing a cross-functional risk management team that includes subject-matter experts (e.g., doctors and nurses) and representatives from research, marketing, engineering, quality assurance, regulatory affairs, and clinical affairs departments. Before the plan is put into effect, the team should establish criteria for categorizing the severity of each potential harm, its likelihood of occurrence, the effectiveness of design or process controls, and risk priority.
Choosing an appropriate methodology for assessing risk will help organize the team's activities. Such methodologies include preliminary hazard analysis (PHA); failure mode and effects analysis (FMEA); failure mode, effects, and criticality analysis (FMECA); fault tree analysis (FTA); and hazard analysis and critical control points (HACCP). Each methodology has different strengths and should be chosen with consideration for the phase of product development, the level of concern for product safety, and the amount of detail required. The output of any of these methods should include prioritizing the risks that were identified and recommending steps to reduce and control those risks.
For example, a device manufacturer can use several techniques to aid in risk assessment. A PHA conducted during the design input phase of development can be used to generate safety-related requirements. It can also indicate areas where the design is not well understood and can recommend further analysis using FTA. During the design verification phase, critical components may require detailed analyses from an FMEA to ensure that all significant risks are reduced or eliminated. All of these techniques together support a risk management effort.
 Figure 4. Risk regions.
Figure 4. Risk regions.
As defined earlier, risk is a two-dimensional measure based on the severity of potential harm and its likelihood of occurrence. FDA and ISO guidelines are represented in the conceptual illustration shown in Figure 4.6,7 In this greatly simplified figure, there are three risk regions; in actual practice, more regions may be defined. A company's goal is to assign hazards to the risk regions to help determine whether risk reduction actions are necessary. ALARP, the acronym in the middle region, stands for as low as reasonably practicable. If the hazard falls in this middle region, then "risks are reduced to the lowest level practicable, bearing in mind the benefits of accepting the risk and the cost of further reduction."7 As a hazard approaches the upper boundaries of the ALARP region, more effort and resources may be needed to reduce it. Safety and efficacy are always the key criteria for determining whether risk has been reduced to an acceptable level.
 Figure 5. FDA and ISO view of the risk assessment process.
Figure 5. FDA and ISO view of the risk assessment process.
The steps for a risk assessment process, illustrated in Figure 5, are described in FDA and ISO guidelines.6,7 The figure shows a waterfall-like process, although in some cases iteration may be necessary before a device can be considered safe. Any change in a device's design or intended use will necessitate a risk assessment review. If testing uncovers an unanticipated product nonconformance to specifications, the effect on risk should be evaluated. Likewise, risk must be assessed in the event of safety-related complaints or returned products.
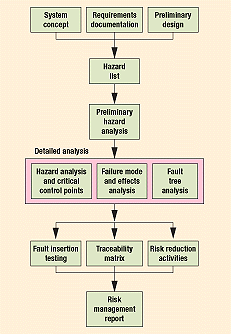 Figure 6. A risk management process.5
Figure 6. A risk management process.5
So how does all of this fit together? Figure 6 outlines a risk management process that blends all of these activities into a logical flow. It matches the product development flow from the conceptual phase to product release. Data collection and a PHA can be accomplished just after the requirements development phase and before a detailed design is established. Detailed analyses, if necessary, can be carried out during detailed design and production process development.
These analyses identify safety requirements, which are part of design inputs. They also yield recommendations for risk reduction, part of design outputs. During the testing phase, design or process controls should be traced from requirements to design to test in order to ensure closure on all risk-related issues, as part of design verification. Fault insertion tests, product tests, process validation, clinical trials, and beta-site tests are all techniques to help gauge the effectiveness of risk reduction measures and are part of design validation. Finally, a risk management report should be issued before design transfer and product release.
CONCLUSION
A well-integrated risk management process offers a manufacturer many benefits. As a "before-the-event" activity, it can discover design inadequacies early in product development, avoiding costly changes or surprises during the test phase. It can also minimize the chances for physical injury and safety-related product recalls and provide some liability protection by acting as evidence of due care.
Risk management also offers benefits that are not as measurable. The use of cross-functional teams can foster cooperation and improve morale—team members feel that they are contributing directly to the product's success. The teams can also improve communication throughout the company—ideas are exchanged that otherwise may never have crossed over organizational boundaries. Finally, the risk management team can be proud of a device that improves the safety of healthcare delivery.
REFERENCES
1. Current Good Manufacturing Practices (cGMP) Final Rule, Supplementary Information, Federal Register, 61 FR:52602.
2. "Design Control Guidance for Medical Device Manufacturers," Rockville, MD, FDA, March 1997.
3. ISO/DIS 14971, "Medical Devices—Risk Management—Part 1: Application of Risk Analysis to Medical Devices," Geneva, International Organization for Standardization (ISO), March 1996. This standard can be obtained from any member body or directly from the Central Secretariat, ISO, Case postale 56, 1211 Geneva 20, Switzerland.
4. Code of Federal Regulations, 21 CFR 820, October 7, 1996.
5. Knepell PL, "Workshop on Risk Management for Medical Products and Processes," Colorado Springs, CO, Logicon RDA, 1997.
6. "ODE Guidance for the Content of Premarket Submission for Medical Devices Containing Software" (draft version 1.3), Rockville, MD, FDA, August 12, 1996.
7. ISO 601-1-4, "Medical Electrical Equipment, Part 1: General Requirements for Safety, 4. Collateral Standard: Programmable Electrical Medical Systems," Geneva, ISO, 1996.
Peter L. Knepell, PhD, is president of Peak Quality Services (Colorado Springs, CO) and formerly the director of software quality and risk management services for Logicon RDA (Colorado Springs, CO).
Copyright ©1998 Medical Device & Diagnostic Industry
You May Also Like


