Sign up for the QMED & MD+DI Daily newsletter.
Integrating Risk Management into the Design and Development Process
Originally Published March 2001Meeting the regulatory requirements for risk management means starting early in the design process and managing risks throughout a product's life cycle.Andrew Snow
March 1, 2001
20 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Integrating Risk Management into the Design and Development Process")
Originally Published March 2001
Andrew Snow
 Successful risk management is essential to the design and development of safe and effective medical devices. Unfortunately, it is too often viewed by manufacturers as an isolated activity that must be performed merely to fulfill a regulatory requirement. This is the result of three common mistakes: a misunderstanding of the regulatory requirement, confusion about what risk management really is, and a failure to recognize the benefits of effective risk management.
Successful risk management is essential to the design and development of safe and effective medical devices. Unfortunately, it is too often viewed by manufacturers as an isolated activity that must be performed merely to fulfill a regulatory requirement. This is the result of three common mistakes: a misunderstanding of the regulatory requirement, confusion about what risk management really is, and a failure to recognize the benefits of effective risk management.
This article clarifies the regulatory expectations and explains the fundamental concepts of risk management. Reviewing the methods of risk management provides manufacturers with tools that will improve their design and development efforts. The end results include device designs that are safe and effective, a shorter and more-efficient design and development timeline, and fewer postlaunch problems.
When it is performed correctly, risk management involves the development and transfer of safe, reliable, and effective devices to manufacturing, while at the same time reducing, controlling, and monitoring risk throughout a device's life cycle. Fundamental to achieving this success is integrating the methods of risk management into the design and development effort so that the following occur:
Risk management becomes part of the seamless flow of design and development.
Risk management evolves with the device design.
The application of design controls is commensurate with the risk associated with the device.
The links between hazards, requirements (and associated design outputs), and verification and validation testing are complete and easily traceable.
Risk management processes form a foundation for decisions regarding the acceptability of device safety and efficacy.
Risk assessments, reductions, controls, and monitoring are transferred as part of the design output to ensure risk management throughout the life of the device.
REGULATORY EXPECTATIONS
To understand how to achieve these results, it is necessary to understand the regulatory expectations for risk management—what it really means and how to do it. This is best achieved by first defining the regulatory requirements for risk management, and then exploring methods for establishing a successful risk management process.
Risk has always been an important part of regulatory requirements for devices, and manufacturers must realize that risk underlies the basic flexibility of the quality system regulation (QSR). Manufacturers have to decide how to implement QSR within their organizations. The preamble provides the guiding principle that the type and extent of controls implemented must be commensurate with the risk associated with the product produced.1 Nowhere is this principle truer than with design controls.
Risk analysis is specifically mentioned in QSR part 820.30(g), Design Validation.2 This part of the regulation states that design validation "shall include software validation and risk analysis, where appropriate." Requirements qualified with "where appropriate" are deemed necessary and appropriate unless the manufacturer can justify otherwise. That is easy enough for software—if there is no software, software validation is not appropriate. In order to justify not doing risk analysis in other cases, however, the manufacturer needs to establish that there are no risks; to do that, the manufacturer needs to perform a risk analysis. Clearly, it is always appropriate to do some form of risk analysis.
This is the area where a narrow interpretation of the regulation can cause confusion. To that end, it is unfortunate that the term risk analysis is buried way down in part 820.30(g) of the regulation, which further contributes to a common misunderstanding of the term. It is tempting for many manufacturers to think that as long as a risk analysis is in the file, all expectations have been met. This is incorrect. To understand why, one must take a closer look at what is meant by risk analysis.
First, the term risk analysis is confusing when placed in the context of current global risk management standards. However, at the time the regulation was developed, it was the term of choice and encompassed all of the activities now understood as risk management; that is, that companies should identify hazards, estimate risks, evaluate the acceptability of risks, and, where unacceptable, implement measures to control those risks and verify their effectiveness.3 In addition, when design changes are made, manufacturers need to evaluate their effects on any existing risk, and then determine if new hazards have been introduced as a result of those changes.
Second, risk management is defined in the standards as a lifecycle activity that starts—at the latest—when design control begins. FDA agrees: in the design control section of QSIT, the agency asks for clarification on how a company managed its risks during the design and development phase of the project.4 FDA wants to see how risk management activities were addressed in design plans and how risk was considered throughout the design process.
This approach is consistent with ISO 13485, the application of ISO 9001 to medical devices, where risk analysis is mentioned in the general requirements for design controls (section 4.4.1).5 "Throughout the design process," the requirement states, "the supplier shall evaluate the need for risk analysis." That is, some form of risk management is always required, it should be addressed as part of the design plans, and it must be considered throughout each stage of the product development process. This means that risk management outputs help define safety requirements as part of design inputs. The risk outputs also determine failure modes to be considered during design validation and identify potential new risks that might result from design changes.
Underlying these requirements is the notion that the types and extent of design controls used for any one product should be commensurate with the risk and complexity of the design. This is an important concept, because the largest potential source of failures associated with design are systematic errors in the design process. Examples of systematic errors would include a failure by the manufacturer to consider or properly identify requirements, the selection of inadequate designs, or a failure to adequately verify or validate. One of the primary benefits of implementing design controls is preventing these types of errors.
Manufacturers often protest that they don't need specific risk analysis techniques, using the all-too-common thought process that because they always take risk into consideration during product design, risk management simply becomes a natural part of the process. But while manufacturers may claim that it is their intention to always consider risk, they do not always follow through on that intent.
Ensuring that risk is considered is one of the major benefits of risk management. It provides a framework within the design control process for reducing systematic error and creates a decision-making process for assessing the adequacy of design safety; when taken together these factors lead to design improvements. In order to see how to integrate this concept into the design and development process, however, a better understanding of the risk management process is required.
RISK MANAGEMENT PROCESS
Risk management is more about the process than it is about the outputs. The documented results must be accurate, complete, and conclusive; more importantly, however, the manufacturer must be able to show how those outputs were used to drive the design control process and create a safe design. Too often, risk management outputs are placed in a design history file and forgotten. Instead, the risk management process should reflect the evolution of a safer device as the design progresses from concept to production.
The fundamental concepts of risk require the recognition that there is a causal relationship among the harm, the hazard, and the cause of the hazard. The cause may occur in the absence of failure or as a result of one or more failure conditions. Often, the hazard is inherent in the nature of the product. Attempting to overcorrect a hazardous event, however, may create further adverse consequences. Therefore, it is the source of the harm and not the actual harm itself that must be dealt with. During risk analysis, both the severity of the consequences of the hazard and the probability of the hazard occuring are evaluated. These two components— severity and probability of occurrence—make up risk.
Often, many of the risks associated with a product are related to the requirements and benefits derived from the product itself; the hazards and risks must then be weighed against the benefits derived. This evaluation determines the acceptability of the risks: if the risk is unacceptable, mitigations or risk reduction measures can be implemented. Doing so, however, might alter the end product negatively, or worse, may create new potential hazards. This back and forth process—assessing risk and reducing it, then evaluating new risk against the benefits derived—is the essence of risk management.
The most widely recognized standard for risk management is ISO 14971.6 It makes a clear distinction between risk analysis and risk management. While this standard is not mandated by the quality system regulations, and there are others (e.g., IEC 60601-1-4, for programmable medical electrical equipment, and EN 1441), it applies to all types of devices and has the common elements required for effective risk management. For a thorough review of the risk management process one can rely primarily on the revised version of ISO 14971.7
Risk analysis is only one element of the risk management process. Figure 1 shows the overall process for risk management. It comprises five core activities: planning, risk analysis, risk evaluation, risk control, and postproduction control. The process begins with planning, and is followed immediately by risk analysis. When the risks are evaluated and decisions are made regarding their acceptability, risk assessment is complete. Risk management begins when risk controls are implemented, their effectiveness is verified, and the overall safety of the device design is deemed acceptable.
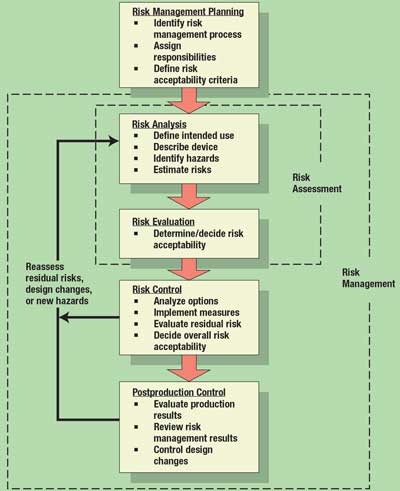
Figure 1. Overall risk management process. Adapted from ISO 14971.
The process of risk management is never really completed; manufacturers must continue to review risk management information as field experience is gained and postproduction design changes are made. This process continues for as long as the product is on the market. The process loops back to evaluate new hazards as design changes are implemented during the postproduction period.
Risk Management Planning. Clearly, the first step in the process is planning how to manage risk for a given project. A risk management plan should include the following:
The scope of the project—which products and phases of the project the plan covers.
The risk acceptability criteria.
The risk review requirements.
The risk activities and resources.
Verification and validation plans.
The risk management plan should go hand in hand with the design and development plan. At the beginning of a project, the nature of hazards and their causes are often unknown, so the plan may change as more is learned about the device. The plan may be project specific, or it may be specified as part of operating procedures and policies. Risk management activities can be included as part of other design reviews or performed as independent reviews. Ultimately, hazards and their mitigations should link directly to verification and validation plans. It is important for management to determine responsibilities, establish adequate qualified resources, and review risk management activities and results to ensure that an effective management process is in place.
When setting policies for risk acceptability, several factors should be taken into consideration. While the overall guiding principle is that risks should be outweighed by benefits, decisions usually can be justified by doing three things: one, comparing the product to other similar devices on the market; two, following appropriate guidance (e.g., the single-fault philosophy); and three, using product-specific standards.
Comparisons with other products should take into account similarities and differences in intended use, hazards, risk, safety features, and historical data. The single-fault philosophy, detailed in IEC 60513, implies that medical electrical equipment have two means of defense against any one hazard, so that a single fault cannot result in the hazard.8 The underlying assumption is that the equipment is reasonably reliable, so that the probability of any one single failure is low. Product-specific standards usually specify requirements, which, if implemented and tested, will result in an acceptable level of risk.
Risk Analysis. The risk management process continues with a definition of the intended use of the device and detailed description of the characteristics that affect device safety. The statement of intended use should also include foreseeable misuse. Annex A of ISO 14971 provides questions that guide the determination of intended use and characteristics of the device.9 Human factors issues and user interfaces should also be taken into consideration at this point.
Next, hazards associated with the device are identified. Brainstorming is a useful tool for identifying hazards and can be supplemented with Annex D of ISO 14971 and Annex B for in vitro diagnostic devices.10 Requirement documents are also good sources of hazards as there are many hazards associated with the nonfulfillment of a requirement. For example, in dialysis equipment there may be requirements for fluid removal and hazards associated with inadequate or excessive fluid removal.
Estimating the risks associated with each hazard completes the risk analysis part of the process. As previously defined, risk is the probability of a hazard causing harm and the severity of the consequences. Risk estimation can be quantitative or qualitative. Some typical tools for risk analysis are summarized in Table I. Risk can also be defined using the chart shown in Figure 2.11 This chart can be converted to a table for a more qualitative assignment of risk based on categories.
Technique | Description | Applications |
|---|---|---|
Preliminary hazard or risk analysis (PHA or PRA) | An inductive method for identifying hazards and estimating risk based on intended use and a description of the device characteristics. Identifies hazards and estimates risks by assigning severity ratings to the consequences of hazards and likelihood-of-occurrence ratings to causes. Can be initiated with conceptual information and updated as design evolves. Helps to identify risk reduction measures early to help define requirements and test plans, and therefore helps ensure design controls for a project are commensurate with risks. | Hazard identificationInitial risk estimatesRequirements definitionCan be adopted for risk management summary reportsInput for project planning |
Fault tree analysis (FTA) | A quantitative method for identifying the potential causes of a hazard or some other specified undesired event (top-level event). Starting with the top event, causes or failure modes are deduced from lower-level systems and component functions and specifications. The causal events can be related to software, hardware, mechanical, or human sources. The output is a fault tree showing the logical relationship of failure events. It helps to identify single-point failures, any one of which may result in the top-level event, or multiple failure conditions where two or more events must occur for the top-level event to occur. The probability of the top event can be predicted using estimates of failure rates for individual events or failure. Failure path sets can be identified to help indicate which events are major contributors to the top event. | System risk analysisSoftware risk analysisEvaluating the effectiveness of process control plansEvaluating processequipment failures/reliability |
Failure modes and effects analysis | An inductive technique that systematically analyzes design or process functions and determines failure modes, their causes, and effects. Risk is estimated by rating the severity of failure effects, the likelihood of causes, and the likelihood of detecting the cause of failure or the failure mode. Can be used to identify potential risk reduction measures and estimate their effects on risk. Typically used as a bottom-up approach starting with components and using a single-point failure approach to progressively work up to the top level. Can be readily adapted to evaluating human error in use applications by using the process FMEA approach. | Component risk analysisProcess risk analysisUse application risk analysis |
Failure modes effects and criticality analysis | Uses the same basic concepts of FMEA, however the output is very different. The method determines the failure rate of a specific component for a given system-failure severity category. The calculation is based on evaluating for each failure mode the conditional robability of the failure effects occurring given that the failure mode occurs, the probability of the component failing in the identified mode, and the component failure rate adjusted for differences between the operating and environmental stresses under which the failure rate was measured and the actual use conditions. It therefore relies on extensive knowledge and data to support failure rates and probability. Data are either based on historical information or extensive testing. Because component criticality is determined, components can be ranked and risk reduction prioritized. It can also be used to develop component specifications. It also helps to develop parameters for use environment. | Component or subassembly risk analysis or reliability analysis. Comparison of alternate designs for components or subassembliesSpecification of operating environments |
Human reliability assessment | Evaluates the influence of human error on the performance of a task. The objective is to estimate the likelihood of the task being performed correctly. It focuses on errors related to the operator not performing a required action, not performing the action correctly, or performing an action that was not required. The technique also evaluates the opportunity and likelihood of error recovery. There are three basic steps: task analysis, error identification, and reliability quantification. Task analysisdescribes and characterizes the nature of the tasks, their sequence, timing, tools available, personnel qualifications, and the environment. Error identification analyzes the sequence of events that result in the error and the consequences of the error. An event or fault tree may be used to help describe the sequence and include recovery events. This is quantified by assigning probability to determine the likelihood of the task being performed error free (reliably). | Human factors analysisIdentifying user interface requirements and instructions for use |
Table I. Survey of common risk analysis tools.

Figure 2. Risk acceptability chart.
Manufacturers should develop a qualitative categorization of severity based on an evaluation of both the long-term and short-term effects of harm. Typically, several discrete levels are developed (see Table II, which was adopted from IEC 60601-1-4).12
Symbol | Descriptor | Description |
|---|---|---|
S1 | Negligible | Little or no potential of injury |
S2 | Marginal | Potential of injury |
S3 | Critical | Potential of death or serious injury |
S4 | Catastrophic | Multiple deaths or serious injury |
Table II. Qualitative severity categories.
If sufficient data are available, quantitative probabilities of hazards can be estimated. Frequently this is not the case, however, and qualitative descriptors (e.g., incredible, improbable, remote, occasional, frequent) must be used to define probability. In estimating probabilities, the manufacturer needs to consider the initiating causes of a hazard, and decide if the hazard occurs in the absence of a failure or from a single or multiple failure. To help estimate this, historical data, analytical or simulation techniques, and expert judgment can all be used. Historical data or simulation techniques are preferable and can act as independent checks of each other. In some cases, however, it may only be possible to use expert judgment.
Risk Evaluation. Once the components of risk have been determined, evaluating the risks associated with each hazard and determining their acceptability completes the risk assessment. Tables III and IV—which are adopted from Annex E in ISO 14971 and IEC 60513, Technical Report on the Fundamental Aspects of Safety Standards for Medical Electrical Equipment—illustrate the decision criteria.13
Severity
Probability | Frequent | R3 | R4 | R4 |
|---|---|---|---|---|
Probable | R2 | R3 | R4 | R4 |
Occasional | R2 | R2 | R3 | R4 |
Remote | R1 | R2 | R2 | R3 |
Improbable | R1 | R1 | R2 | R2 |
Incredible | R1 | R1 | R1 | R1 |
Table III. Qualitative risk categories.
Risk Level | Acceptability |
|---|---|
R1 | Broadly acceptable, no need to consider further risk reduction |
R2 | Risk is unacceptable and should be reduced as low as reasonably practicable (ALARP); technical and economic practicability are balanced against risks/benefits |
R3 | Risk is unacceptable and should be reduced as low as reasonably practicable (ALARP); technical practicability is balanced against risks/benefits and risk is reduced even at considerable costs |
R4 | Intolerable, risk is unacceptable and must be reduced |
Table IV. Risk acceptability.
Generally, risks that are as low as reasonably practicable (ALARP) are acceptable if the benefits justify any residual risks. At the same time, however, intolerable risks are not acceptable and must be reduced at least to the level of ALARP risks. If this is not possible, the project must be terminated.
The concept of practicability involves both technical and economic consideration. Technical in this case refers to the availability and feasibility of solutions that mitigate or reduce risk; economic refers to an ability to reduce risks without making the use and application of the device financially infeasible.
If the risk is not broadly acceptable, risk reduction must be considered. There are three possible scenarios in this case: one, risk is reduced to the broadly acceptable region and there is no need to consider it further; two, risk reduction is practicable, but it cannot be reduced into the broadly acceptable region; or three, risk reduction is not reasonably practicable.
In the latter two scenarios, residual unacceptable risk is evaluated against the device benefits to determine its acceptability. Benefits should include consideration of the patient's current state and prognosis, the likelihood of improvement or deterioration if alternative treatments are used, and the degree and likelihood of improvement with the proposed treatment. If the benefits do not outweigh the risks, the risk is unacceptable.
Risk Control. Once the decision is made to reduce risk, control activities begin. Risk reduction should focus on reducing the hazard severity, the probability of occurrence, or both. Manufacturers should use one or more of the following types of risk controls, in the order listed:
Inherent safety by design (e.g., a more robust design or a design with greater safety margins).
Protective design measures (fail-safes, warnings, or alarms).
Protective manufacturing measures, which improve process or test capabilities.
Information for safety (labeling, instructions for use, training, etc.).
Product-specific standards address inherent safety, protective measures, and information for safety (e.g., labeling) for many types of devices. These standards should be used to define requirements for design and testing, where applicable. One way of evaluating the control options is to estimate their potential impact on the severity and probability of hazard occurrences. The technical and economic practicality of implementing the options should also be evaluated. If it is not practicable to reduce risk further, then the risk/benefit analysis must justify any residual risks.
An implementation of risk reduction measures consists of the following:
Implementing the design requirements.
Verifying effectiveness.
Assessing potential new hazards and risks.
Evaluating any residual risks.
Once the risk reduction measures have been fully evaluated and implemented, the overall risk acceptability of the device should be determined. This is done using the criteria established in the risk management plan (which is based on individual hazards). The risk acceptability evaluation should be documented in the final risk management report. The risk management report contains or refers to the following:
The device's intended use and description.
The risk methods used.
The risk acceptability criteria selected.
All identified hazards.
The risk estimations.
The risk controls.
A verification of control or mitigation effectiveness.
Evaluations of residual risks for individual and overall device safety.
You May Also Like


