FDA approved Boston Scientific's Farapulse system in January for pulsed field ablation treatment of atrial fibrillation.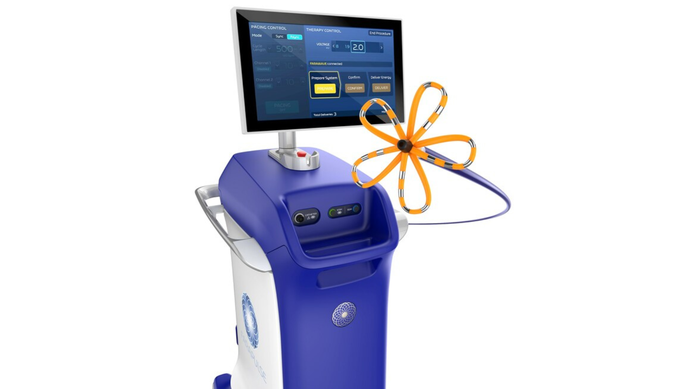
CardiovascularBoston Scientific's Farapulse Is a ShowstopperBoston Scientific's Farapulse Is a Showstopper
Electrophysiologists are giving the new pulsed field ablation system a warm welcome.












.png?width=300&auto=webp&quality=80&disable=upscale)
















.png?width=300&auto=webp&quality=80&disable=upscale "NeuroStar")

.png?width=300&auto=webp&quality=80&disable=upscale "Siemens Healthineers")
Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.