Bethany Corbin Women's Health Thought Leader
ManufacturingFemInnovation CEO to Keynote IME SouthFemInnovation CEO to Keynote IME South
Bethany Corbin, a recognized thought leader in women's health, is set to keynote IME South. The six-in-one advanced design and manufacturing expo returns June 4-6 to the Charlotte Convention Center.




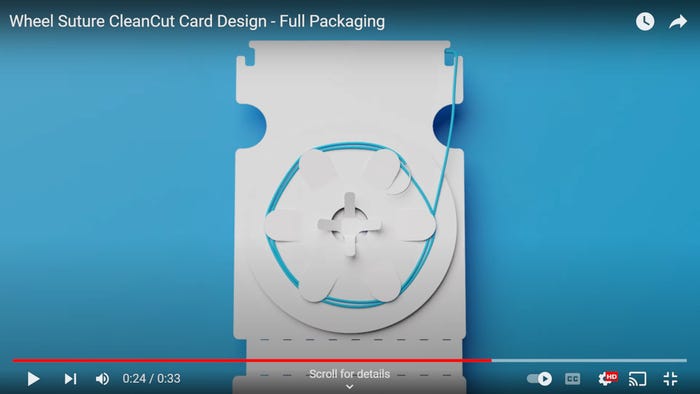


 Closure device.")

.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "Medical sterilization")




.png?width=300&auto=webp&quality=80&disable=upscale "Global supply chain")








.png?width=300&auto=webp&quality=80&disable=upscale "Intricon")


.png?width=300&auto=webp&quality=80&disable=upscale "Freudenberg Medical")
.png?width=300&auto=webp&quality=80&disable=upscale "medical device sterilization")
.png?width=300&auto=webp&quality=80&disable=upscale "Medical Molding Outsourcing")


.png?width=300&auto=webp&quality=80&disable=upscale "A photo of the Anaheim Convention Center")






Editors' Choice


.png?width=300&auto=webp&quality=80&disable=upscale)
Sponsored Content
May 23, 2024
Sign up for the QMED & MD+DI Daily newsletter.