Sign up for the QMED & MD+DI Daily newsletter.
Software-related recalls have jumped 111% since 2010. Here’s what’s behind the increase.
July 7, 2016
6 Min Read

Software-related recalls have jumped 111% since 2010. Here's what's behind the increase.
Joshua R. Dix, Suraj Ramachandran, Darin S. Oppenheimer
Editor's Note: This is the second installment in a four-part series on the factors behind medtech's recall epidemic. Read Part 1 and Part 3.
Much like the Cooper Committee Legislation of 1970, both FDA's Safe Medical Device Act of 1990 and the 1996 Quality System Requirements were prompted into legislation by a set of tragic events related to the use of medical devices. Between 1985 and 1987, cancer patients undergoing radiation treatment through the Therac 25 Accelerator device were given massive overdoses of radiation due to a concurrent programming error in the device software. The incident, thought of as the worst series of radiation accidents in the 35-year history of the technology, resulted in six adverse events, including multiple instances of severe burns, irreversible limb paralysis, and, catastrophically, the death of two patients.
While accidents like the one described above brought to light the dangers of software-controlled medical devices and prompted health authorities to establish more stringent regulatory controls regarding the design, validation, and manufacture of software, the continuing advancement of technology, coupled with a reactive industry model has led to a vicious cycle of exciting progression followed by an equal and altogether more frustrating regression.
A recent analysis, conducted as part of this research, used CDRH's recall repository to collect data and evaluate trends in both voluntary and involuntary recalls established between January 2010 and December 2015. The product of this evaluation demonstrates an average of approximately 225 software-related recalls annually, according to Blue Lynx Consulting (Figure 1). The data also specifies a progressive yearly occurrence rate with trending indicating an upward swing in the following root causes as identified by the agency: software change control, software manufacturing process design, and software design.
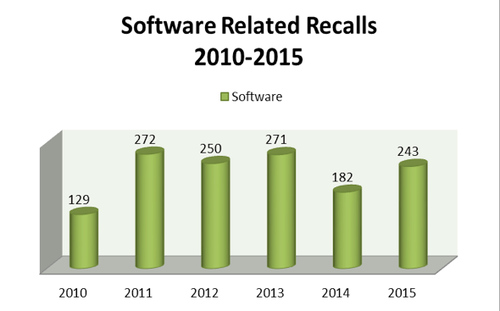
Figure 1. Software recalls by year (raw data), 2010-2015
This article explores the specific recalls, hazardous situations, trends, and possible mitigations related to each of the three root causes FDA directly correlated to a five-year upward trend in software-related medical device recalls.
Software Recalls
While software controlled devices predate the 1976 Food, Drug, and Cosmetic Act officially expanding FDA's scope of jurisdiction to medical devices, the ever-escalating complexity of software technology continues to be a challenge for health authorities. As evidenced by the release of both the 2002 General Principles of Software Validation and 2005 Content of Premarket Submission for Software Controlled Medical Devices guidance documents, FDA has initiated "ongoing efforts to state their recommendations more clearly and ensure they remain current as technology advances." In addition, 11% of FDA's planned draft guidance documents for 2016 are specific to the topic of software. Despite these continuous improvement efforts, recalls related to the control of software account for nearly 8% of the total number of recalls since 2010, according to Blue Lynx Consulting.
In addition to being a prevalent cause of medical device recalls, software-related recalls are also rising steadily, accounting for a 111% cumulative increase since 2010. Unlike other root causes of device issues, software-related recalls are unique in the sense that each of the three subcategories has also shown a steep and steady growth. As demonstrated in Figure 2, software design (+96%) software change control (+214%), and software process design (+460%) have each shown an implausible upward trend in the past five years.
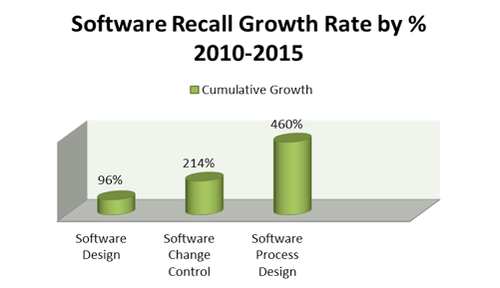
Figure 2. Recall growth rate percet, 2010-2015
Even after the industry has had ample time to digest guidance documents such as such as FDA's 2002 General Principles of Software Validation, the upswing of death and serious injuries related to design issues continue. As evidenced by the upward trend of recalls and the multiplicity among those companies that experience recalls for the same reasons annually, it is clear device companies still struggle to identify proper mitigations for the reoccurrence of these hazards.
Why Do Issues Reoccur?
Despite FDA's best efforts to stay ahead of technological complications posed by an influx of intricate software controlled devices, analytics of recall trends related to software continue to demonstrate a disconnect between the availability of guidance and risk information to the manufacturer and the ability of the manufacturer to actually mitigate issues, according to Blue Lynx Consulting (Figure 3).
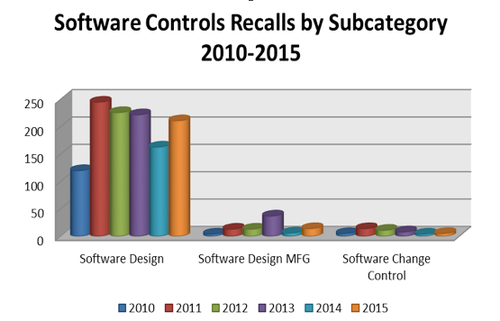
Figure 3. Software-control-related annual recalls by subcategory, 2010-2015
It is apparent that FDA is investing considerable time and effort in trying to reduce device problems and recalls; it is also clear that regulations for manufacturers incorporating risk management have been in place for several years. What is not clear is how device manufacturers are implementing these requirements. Therefore, we must again ask whether the information provided as part of this writing indicates a problem that is more behavioral than technological.
As we move into the next part of this series, we will explore the recall trends related to production controls and begin to further discuss the questions surrounding how medical device companies can counteract the current negative trends.
Disclaimer: The perspectives shared in this article represent the personal opinions and insights of the individual authors and are not associated with the authors' respective employers.
Note: All supporting statistical information garnered from the FDA Recall Database in support of this article was compiled and provided by Blue Lynx Consulting.
Joshua R. Dix is a regulatory affairs specialist. Based in the Western New York area, he is the global regulatory lead for multiple product platforms with responsibilities including regulatory strategy, submissions, product CAPA, and audits. With extensive experience in both regulatory affairs and quality systems, Dix has worked to bring multiple medical devices to market in more than 20 different countries. He holds a bachelor's degree in English from the State University of New York.
Suraj Ramachandran, MS, is a regulatory affairs manager. Based in the Chicago area, he is involved primarily with managing the infusion pump platform and supporting all new product development and life cycle maintenance activities, including regulatory submissions, design control, audits, and CAPAs. Ramachandran has also led many development efforts regarding medical device software intended for both domestic and international markets. He holds an undergraduate degree in biomedical engineering as well as a master's degree in biomedical engineering from the University of Michigan. In addition, he holds a RAC certification from the Regulatory Affairs Professional Society.
Darin Oppenheimer, MS, is a director of regulatory affairs. He is involved in many facets of the product development life cycle, including regulatory submissions, due diligence, and active participation on industry trade organizations and standards committees. Oppenheimer leads a team of regulatory professionals focusing on electromechanical devices and software. His prior background as a research and development scientist focused on pharmaceuticals and medical device diagnostic applications for biomarker and drug discovery. His undergraduate degree is in molecular biology from the University of Tampa. He also holds two master's degrees from Johns Hopkins University in biotechnology and regulatory science, as well as a graduate certificate in biotechnology enterprise.
[image courtesy of NIYAZZ/iSTOCKPHOTO.COM]
You May Also Like


