Sign up for the QMED & MD+DI Daily newsletter.
FDA cited device-design issues as one reason medical device recalls are on the rise. Here’s how manufacturers can stop these problems from reoccurring.
June 27, 2016
8 Min Read

FDA cited device-design issues as one reason medical device recalls are on the rise. Here's how manufacturers can stop these problems from reoccurring.
Joshua R. Dix, Suraj Ramachandran, and Darin S. Oppenheimer
Editor's Note: This is the first installment in a four-part series on the factors behind medtech's recall epidemic. Read Part 2 and Part 3.
Since the 1970 Cooper Committee Report documented thousands of deaths and serious injuries related to the use of medical devices, FDA has taken a focused approach in its microscopic tracking of medical device adverse events and recalls, culminating in the October 2014 issuance of the guidance Distinguishing Medical Device Recalls from Medical Device Enhancements. As advancements in technology have provided the agency with greater resources to compile and communicate data to industry and consumers alike, reports of events like those first demonstrated in the Cooper Report have become more frequent and progressively alarming.
At the conclusion of 2006, FDA communicated a staggering set of data to the medical device community. A calendar year's worth of agency data was conclusive in attributing 116,000 injuries, 96,000 malfunctions, and 4500 deaths to the use--or, in this case, misuse--of medical devices. Further analysis released in 2012 in the Medical Device Recall Report FY 2003 to FY 2012 reported a 97% increase in recalls from the fiscal year (FY) 2003 (604 total) to the FY 2012 (1190 total).
A recent analysis conducted for this article used CDRH's recall repository to collect data and evaluate trends in both voluntary and involuntary recalls established between January 2010 and December 2015. The product of this evaluation demonstrates an average of approximately 2600 recalls annually (See Figure 1), according to ongoing analysis if medical device recalls provided by Blue Lynx Consulting. The data also specifies a progressive yearly occurrence rate with trending indicating an upward swing in the following root causes as identified by the agency: device design, software controls, and production controls.

Figure 1. Recalls by Year 2010-2014
This article is the first in a four-part series that will explore the specific recalls, hazardous situations, trends, and possible mitigations related to each of the three root causes FDA directly correlated to a five-year upward trend in medical device recalls.
Device Design
Since 2010, issues with device design have accounted for nearly 35% of all recall root causes determined upon firm or FDA investigation. Included in this analysis are root causes determined to be directly correlated to subcategories of device design, such as component design, labeling design, packaging design, process design, and software design. While the five-year analysis of design-related recalls does not necessarily demonstrate a consistent upward trend, the mean number of recalls per year documented in this investigation hovers well above 900, indicating continued recall issues across multiple medical device companies and platforms.
While issues surrounding device design are the most prevalent cause of medical device recalls, device-design-related recalls are also the most likely to cause serious health problems or death to the end user. Recalls correlated to device design encapsulate close to 46%of the five-year total of Class I recalls, with a 303% increase between FY 2013 and FY 2014 (Figure 2), according to Blue Lynx consulting. (Note: 2015 data provided by the FDA is incomplete, therefore growth between 2013 and 2014 is the latest information available.)
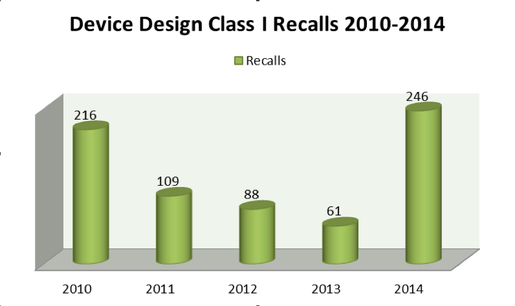
Figure 2. Device-Design-Related Class I Recalls 2010-2014
Despite the proliferation of more complex guidance documents such as the agency's 2002 General Principles of Software Validation, the upswing of death and serious injuries related to design issues continues. As evidenced by the upward trend of recalls and the multiplicity among those companies that experience recalls for the same reasons annually, it is clear that device companies still struggle to identify proper mitigations for the reoccurrence of these hazards. Information collected from the agency's database by Blue Lynx Consulting shows 30% of device companies that instituted a recall for device designs in 2010 were once again opening recalls in 2014 for similar reasons.
Why Do Device-Design Issues Reoccur?
To understand why companies struggle with device design, it is imperative to recognize the various elements of device design and where companies continually fall short. In correlation with the upward trending of device design recalls, medical device manufacturers have failed to consistently mitigate hazards associated with component design, labeling design, packaging design, process design, and software design. Each of these subcomponents of device design have seen a drastic upward trend in related recalls over the past five years, with only component (-51%) and software design (+70%) staying under a 100% growth-rate average over the same five year span.
As demonstrated by Figure 3, companies have struggled immensely over the past five years in their attempts to mitigate recalls derived from elements of device design.
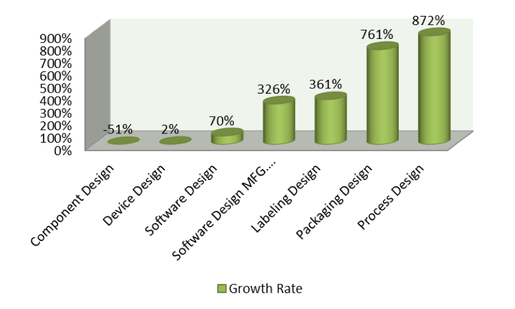
Figure 3. Recall Growth Rate % 2010-2014
Specifically, manufacturers struggled most profoundly in the areas of labeling design (+361%), packaging design (+761%), and process design (+872%) (Figure 4).
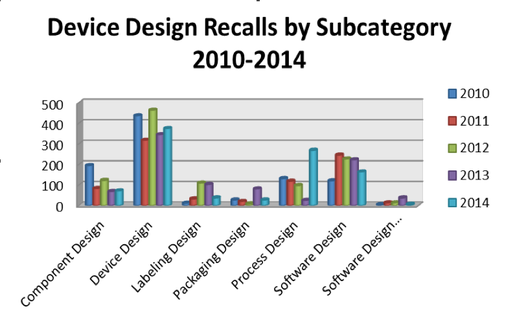
Figure 4. Device Design Related Annual Recalls by Subcategory 2010-2014
Although each of these subcomponents can be tied back to device design, their unique complications should be viewed as impediments for the mitigations of the remaining failures and hazardous situations. While each of these areas of device design come with associated regulations, guidance, and standards, the current reactive state of medical device companies does not allow for continuous growth over a wide variety of situations where the issues are both unique and complex. As noted in Francisco Polidoro Jr.'s 2016 Harvard Business Review article, "Why Do Organizations Forget What They Learn":
The occurrence of a serious error such as a shuttle explosion, a large-scale oil spill, or a safety-related product recall triggers the learning cycle. It stimulates an organization to emphasize safety as a primary concern, as it seeks to identify the root causes and to make the corresponding corrections in its processes, structure and culture. The problem is that these safety-related behaviors fade over time and other motivating forces come to the fore, gradually launching the seeds of the next error.
Using this ideal in the context of a company or industry facing multiple complex challenges concurrently, it is logical to attribute the fluctuation of these recall issues to companies that are unable to mitigate every known issue at once. This is to say that when a company or industry focuses heavily on an upward trend of one issue, it is simply "launching the seeds" for the next upward trend in another area.
While the information and technologies available to the medical device industry have grown exponentially since the days of the Cooper Committee, analytics of recall trends related to device design demonstrate a disconnect in the parallel between availability and use. Does the information provided as part of this article point to a problem that is more behavioral than technological? One has to wonder if the Cooper Committee was conducted at present day, would it drive additional reform in the area of device design based on the reoccurring number of recalls and complaints?
In the next part of this series, we will focus on the recall trends related to software controls and further explore the questions surrounding how medical device companies can counteract the current negative trends.
Disclaimer: The perspectives shared in this article represent the personal opinions and insights of the individual authors and are not associated with the authors' respective employers.
Note: All supporting statistical information garnered from the FDA Recall Database in support of this article was compiled and provided by Blue Lynx Consulting.
Joshua R. Dix is a regulatory affairs specialist. Based in the Western New York area, he is the global regulatory lead for multiple product platforms with responsibilities including regulatory strategy, submissions, product CAPA, and audits. With extensive experience in both regulatory affairs and quality systems, Dix has worked to bring multiple medical devices to market in more than 20 different countries. He holds a bachelor's degree in English from the State University of New York.
Suraj Ramachandran, MS, is a regulatory affairs manager. Based in the Chicago area, he is involved primarily with managing the infusion pump platform and supporting all new product development and life cycle maintenance activities, including regulatory submissions, design control, audits, and CAPAs. Ramachandran has also led many development efforts regarding medical device software intended for both domestic and international markets. He holds an undergraduate degree in biomedical engineering as well as a master's degree in biomedical engineering from the University of Michigan. In addition, he holds a RAC certification from the Regulatory Affairs Professional Society.
Darin Oppenheimer, MS, is a director of regulatory affairs. He is involved in many facets of the product development life cycle, including regulatory submissions, due diligence, and active participation on industry trade organizations and standards committees. Oppenheimer leads a team of regulatory professionals focusing on electromechanical devices and software. His prior background as a research and development scientist focused on pharmaceuticals and medical device diagnostic applications for biomarker and drug discovery. His undergraduate degree is in molecular biology from the University of Tampa. He also holds two master's degrees from Johns Hopkins University in biotechnology and regulatory science, as well as a graduate certificate in biotechnology enterprise.
[image courtesy of NIYAZZ/iSTOCKPHOTO.COM]
You May Also Like


