Ivenix Infusion Pump LVP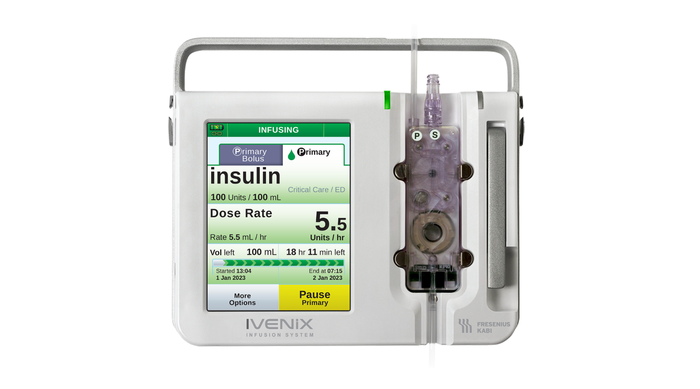
Regulatory & QualityFresenius Kabi USA Infusion Pump Software Recall Declared Class IFresenius Kabi USA Infusion Pump Software Recall Declared Class I
The company is urging customers to update the device software in order to mitigate noted software anomalies.




















.png?width=300&auto=webp&quality=80&disable=upscale "plastic syringe")


.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "EU MDR")
.png?width=300&auto=webp&quality=80&disable=upscale "Axonics R20 Rechargeable Sacral Neuromodulation System")


.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Elon Musk")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA Recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Shane Farritor")

.png?width=300&auto=webp&quality=80&disable=upscale "Regulatory Pathway")

.png?width=300&auto=webp&quality=80&disable=upscale "FDA Safety Communication")
.png?width=300&auto=webp&quality=80&disable=upscale "European Union Council")




Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.