Exo Iris Lung and Cardiac AI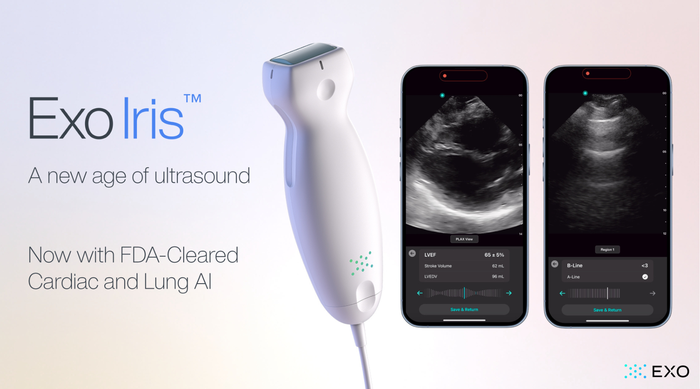
Artificial IntelligenceExo’s FDA Cleared Cardiac & Lung AI Now on Iris Handheld UltrasoundExo’s FDA Cleared Cardiac & Lung AI Now on Iris Handheld Ultrasound
With the newly approved applications, Exo is now cleared for cardiac, lung, bladder, hip, and thyroid.










.png?width=300&auto=webp&quality=80&disable=upscale)

















.png?width=300&auto=webp&quality=80&disable=upscale "NeuroStar")

.png?width=300&auto=webp&quality=80&disable=upscale "Siemens Healthineers")


Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.