

Sign up for the QMED & MD+DI Daily newsletter.
Marcarelli Cooper
+2
12 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Consider This A Warning")
|
|
Helfgott | Cochran |
Eleanor Roosevelt once said: “Learn from the mistakes of others. You can’t live long enough to make them all yourself.” As the first decade of the new millennium comes to a close, an opportunity is presented to learn from mistakes of the past, and embrace progress toward a better future. Apparently, the medical device research community has been learning from its mistakes; the most recent FDA compliance trends indicate that in comparison with previous years, there was a decrease in the number of bioresearch monitoring (BIMO) warning letters (WLs) issued to the regulated device research community. An analysis of FY 2007 WL patterns for the device research community, published in MD+DI’s November 2008 issue under the title “Gleaning Good Practices from Warning Letters,” provides a baseline to make comparisons against this larger collection of WL data.
WLs: A Barometer for the State of Compliance
According to FDA’s Office of Enforcement, the agency issued a total of 471 WLs to regulated industry in FY 2007, 445 WLs in FY 2008, and 473 WLs in FY 2009. In FY 2009 (October 2008–September 2009), BIMO, housed within CDRH, issued 13 WLs. This BIMO total is less than half of the WL totals issued by BIMO in FY 2008 and FY 2007, respectively.
Accordingly, in both FY 2007 and 2008, BIMO WLs accounted for about 7% of the total number of WLs issued by FDA during that time. Such a trend was alarming considering that CDRH BIMO inspections only account for about 3% of the total number of inspections conducted annually by FDA. However, in FY 2009, WLs from BIMO only accounted for 3% of total FDA WLs issued, a dramatic decrease compared with the previous two years. This downward trend may have several causes. It may be due to an increase in the overall observed state of compliance within the device research community. It may also be the result of the regulated device research community’s move toward a quality system approach to the oversight of device clinical trials, as well as BIMO’s best practices outreach and risk-based inspection approaches.1
BIMO Inspection Process
Generally, a field investigator from FDA’s Office of Regulatory Affairs (ORA) conducts the BIMO inspection in response to a written request from CDRH. These inspections focus on verifying the integrity of the data submitted to the agency and ensuring that systems are in place to protect the rights and safety of clinical trial participants. After the BIMO inspection is completed, the ORA field investigator documents inspection observations on FDA Form 483, writes an establishment inspection report, and then provides CDRH with a preliminary classification of the inspection results.2 FDA has three primary inspection classifications:
? Official Action Indicated (OAI): Objectionable conditions were found and one of the regulatory actions (advisory, administrative, or judicial) should be recommended. Typically, an OAI classification should be made only if an FDA-483 has been issued and the documented evidence supports the action recommended.
? Voluntary Action Indicated (VAI): Objectionable conditions were found and documented but the district or center is not prepared to take or recommend any of the regulatory (advisory, administrative, or judicial) actions because the objectionable conditions do not meet the threshold for regulatory action.
? No Action Indicated (NAI): No objectionable conditions or practices were found during the inspection (or the significance of the documented objectionable conditions found does not justify further action).
Significant departures from the regulations or repeated noncompliance can result in the issuance of a WL. FDA’s Regulatory Procedures Manual indicates that a warning letter is informal and advisory in nature.3 It communicates the agency’s position on a matter, but it does not commit FDA to taking enforcement action (e.g., civil money penalties) unless appropriate corrective actions are not taken. FDA’s practice is to give individuals and firms an opportunity to take voluntary and prompt corrective action before it initiates an enforcement action. The use of WLs is based on the expectation that most individuals and firms will voluntarily comply with the law.
Warning Letter Review
CDRH BIMO examined all WLs from FY 2007–2009 issued to device clinical investigators (CIs), sponsor/monitors (S/M), institutional review boards (IRBs), and sponsor/investigators (S/I) to identify compliance trends and risk indicators that could be useful to the device research community as they develop risk management strategies for clinical trials.
|
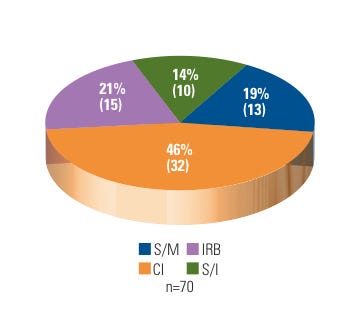
Figure 1. (click to enlarge) Warning letters by firm type. |
During FY 2007–2009, the BIMO unit issued 46% of its WLs to clinical investigators. This is not surprising considering that CDRH allocates almost 60% of its BIMO inspectional resources to the oversight of CIs. Approximately 60% of those CIs inspected were located in academic medical centers or local community hospitals. The remainder were in private practice.
In contrast, 33% of CDRH BIMO WLs went to device research sponsors, to which roughly 12% of BIMO inspectional resources are assigned. The majority of the sponsors were noncommercial or small businesses. In addition, 10 WLs (14%) resulted from OAI follow-up inspections of recidivist entities (recidivist sites are defined as having a history of being in and out of compliance and are often seen as repeat offenders). Fifteen WLs (21%) went to hospital IRBs, of which four were repeat offenders. See Figure 1 for a breakdown of CDRH BIMO WL distribution for FY 2007–2009.
|
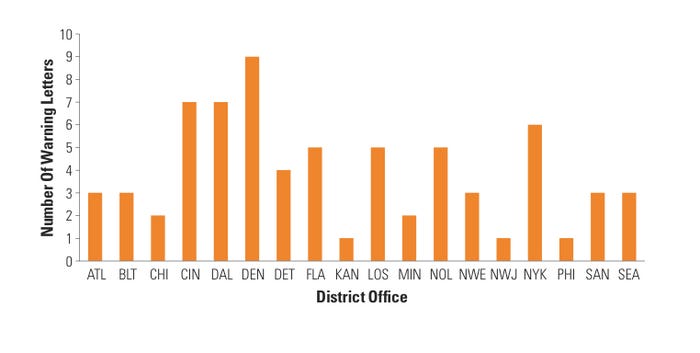
Figure 2. (click to enlarge) Warning letters by district. |
CDRH BIMO also looked at the distribution of device research WLs across the FDA district offices that conducted these inspections. Interestingly, Denver district office inspections resulted in nine warning letters, which accounted for almost 13% of the total number of WLs that CDRH BIMO issued—despite the Denver district receiving only 3% of the device BIMO inspection requests annually. Moreover, five of the 20 FDA district offices (Cincinnati, Denver, Dallas, New Orleans, and New York) accounted for almost half of all the warning letters issued by CDRH BIMO (see Figure 2). These data are again surprising because those five districts generally receive about 25% of all BIMO inspection assignments in a given fiscal year. One explanation may be that CDRH BIMO does a better job of focusing its resources on high-risk device research areas, which may account for the apparent lack of equal geographical WL distribution.
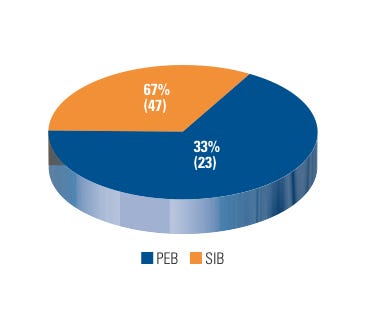
Internally, CDRH BIMO’s Special Investigations Branch (SIB) conducted 47 inspections that resulted in a WL issuance, which accounts for 67% of the total number of WLs issued by CDRH BIMO in FY 2007–2009. CDRH BIMO’s Program Enforcement Branches accounted for the remainder (see Figure 3).
|
Figure 3. (click to enlarge) Warning letters by issuing branch. |
SIB’s primary role is to follow up on complaints of research misconduct and previously violative inspections. The majority of SIB-related WLs were the result of research misconduct complaints, which are typically designated as “for cause” inspections. Exactly half (35 out of 70) of all the WLs issued by CDRH BIMO between FY 2007 and FY 2009 were a result of a “for cause” inspection; other WLs issued were related to repeat offenders and other surveillance inspections.
BIMO’s Program Enforcement Branches issued 23 WLs, primarily due to CDRH’s early intervention and marketing application activities. This total accounts for 33% of the total number of WLs issued by the BIMO unit in FY 2007–2009.
Early intervention activities are risk based and focus on active IDE studies of novel technologies, vulnerable populations, or significant public health issues. It is interesting to note that CDRH BIMO inspections conducted pursuant to marketing applications (i.e., premarket approval, 510(k), etc.) resulted in the issuance of just six warning letters during this period. One explanation is that BIMO’s early intervention program sensitizes device firms to the importance of well-managed clinical trials during the active research phase, leading to marketing applications with higher-quality studies and less-significant compliance shortcomings.
|
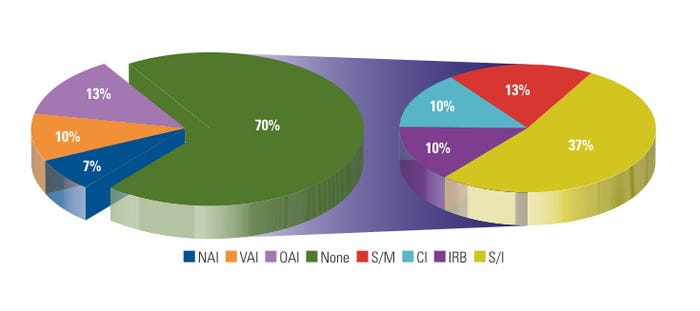
Figure 4. (click to enlarge) Warning letters by previous inspection outcome. |
During FY 2007–2009, CDRH BIMO issued about 15% of its WLs to recidivist sites. In contrast, CDRH as a whole issued 70% of its WLs to sites that were being inspected for the first time during the same period. Figure 4 shows WLs by previous inspection outcome. In most cases, systemic issues such as lack of regulated device research training, lack of management or supervisory oversight, and ineffective corrective and preventive actions (CAPAs) were the primary causes of WL issuance. Some device sponsors of FDA-regulated research exercise risk management by paying increased attention to CIs conducting device research for the first time or CIs that were subject to a prior agency regulatory action. Moreover, the sponsors use various tools and exercises, such as long-distance or on-site training and mock audits, which may assist sites in preparation for a BIMO inspection, and help to avoid possible agency regulatory action.
Compliance Shortcomings
|
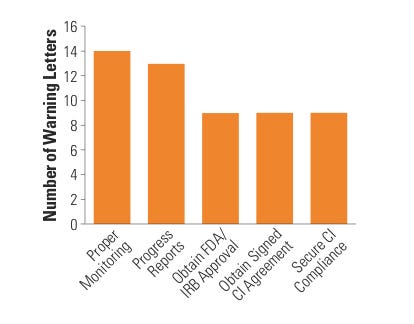
Figure 5. (click to enlarge) Top five sponsor citations. |
The most common compliance problems cited on a sponsor WL include failure to submit an IDE application, supplement, or investigational plan to FDA, investigators, or IRB; inadequate monitoring; failure to obtain FDA or IRB approval; failure to obtain a signed investigator agreement; inadequate device accountability; and inadequate adverse event (AE) or unanticipated adverse device effect (UADE) analysis and reporting (see Figure 5).
The most common compliance shortcomings cited on a CI WL include: failure to follow investigational plan; failure to obtain informed consent; inadequate case histories; inadequate device accountability records; inadequate AE/UADE analysis and reporting; and failure to submit progress reports.
The most common compliance problems cited on an IRB WL include failure to obtain quorum; inadequate written procedures; inadequate minutes; incorrect nonsignificant risk or significant risk (NSR-SR) determination; and conflict of interest.
In addition, CDRH BIMO cited an informed consent shortcoming in 23 of the 70 WLs issued during FY 2007–2009; 20 of the 23 such shortcomings were a result of excluding one of the eight basic elements of informed consent within the informed consent document. Only three WLs contained an informed consent citation in which legally effective informed consent was not obtained.
Trends among WL Responses
Some regulated device research entities have the internal capacity to identify, correct, and prevent their own compliance shortcomings through various mechanisms such as incorporating a quality system approach to the oversight of their research. When a regulated entity receives a WL, the recipient must provide a response within 15 working days of receipt. The objective of a response to a WL is to give individuals and firms an opportunity to take voluntary and prompt corrective action before FDA initiates an enforcement action. The WL response is a critical component of the regulated research entity’s continuous quality improvement process.
Based on our analysis of adequate WL responses, it appears that a previous inspection by FDA has minimal impact on the quality of a regulated entity’s response to an FDA WL. In fact, for sites that FDA inspected for the first time, the response to the WL was adequate 51% of the time, as opposed to entities that FDA inspected previously, for which the WL response was adequate only 48% of the time. Moreover, out of the regulated entities that received a recidivist WL, only 44% responded adequately to it.
Additionally, the most frequent responses to a WL are promises to implement standard operating procedures (SOPs) and to conduct training. We observed a trend among recidivist regulatory violators to implement SOPs but not document training on them. It is important that sites carefully consider the reason for their shortcomings and how new SOPs and training will be able to correct them and prevent them from recurring. Common CAPA strategies include assessing the root cause of the problem, evaluating the extent of the problem, and assessing its impact to ensure that proposed remedies are effective.
|
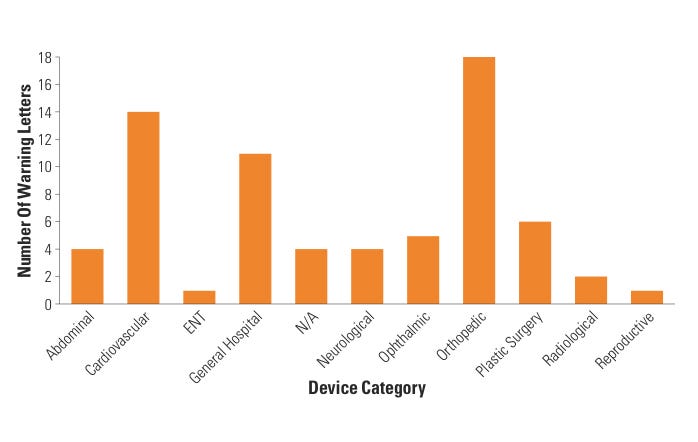
Figure 6. (click to enlarge) Warning letters by device category. |
Lastly, CDRH BIMO looked at specific device categories to assess whether certain device clinical studies tended to generate WLs more than others. During FY 2007–2009, eight categories of devices accounted for all WLs issued by CDRH BIMO, with cardiovascular, orthopedic, general hospital, and plastic surgery device studies topping the list. In fact, almost half of all WLs (46%) came from cardiovascular and orthopedic device–related studies. This outcome was expected because the vast majority of active IDEs fall within the cardiovascular and orthopedic categories. When CDRH stratified the data further, the data revealed that almost half of the CI WLs involved cardiovascular and orthopedic studies. When compared with FY 2007 WL data, it appears that the trend among cardiovascular and orthopedic device categories receiving almost half of all BIMO WLs remains unchanged (see Figure 6).
Conclusion
The regulated device research community could view a WL as an opportunity to improve the quality of its research activities and culture of compliance. Some sponsors and regulated entities of device research incorporate the regulatory shortcomings outlined in the WL into the CAPA component of their in-house quality systems. Such an action may promote continuous quality improvement of research activities, which could lead to higher-quality research data and enhanced human subject protection. Enhanced CAPA systems possess both long-term and short-term benefits such as reduced time to market, less resources dedicated toward continuous monitoring, and a reduction in clinical trial delays.
Additionally, CDRH has seen some device firms successfully use universal risk analysis principles, including failure mode effects analysis and fault-tree analysis, to determine the likelihood of a failure occurring during the clinical trial process and to reduce each posed risk to an acceptable level. These regulated entities do not wait for the issuance of a WL to identify and solve their compliance shortcomings; they have a culture of compliance integrated throughout their total product life cycle. Whatever the method, the device research community seems to have recognized the signal and made a dedicated effort to put its best foot forward.
References
1. M Marcarelli, “Gleaning Good Practices from Warning Letters,” Medical Device + Diagnostic Industry 30, no. 11 (2008). Available from Internet: www.mddionline.com/article/gleaning-good-practices-warning-letters.
2. FDA Inspections, Compliance, Enforcement, and Criminal Investigations. Available from Internet: www.fda.gov/ICECI/Inspections/FieldManagementDirectives/ucm061430.htm.
3. FDA Regulatory Procedures Manual, available from Internet: www.fda.gov/downloads/ICECI/ComplianceManuals/RegulatoryProceduresManual/UCM074324.pdf.
About the Author(s)
You May Also Like


