Sign up for the QMED & MD+DI Daily newsletter.
Medical Device & Diagnostic Industry MagazineMDDI Article IndexOriginally Published May 2000COMBINATION PRODUCTSDevices that also include some functions of drugs or biologics are offering innovative treatment options, but can face hurdles in development.William Loob
William Loob
May 1, 2000
16 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Combination Products Enhance Capabilities, Pose New Challenges")
Medical Device & Diagnostic Industry Magazine
MDDI Article Index
Originally Published May 2000
COMBINATION PRODUCTS
Devices that also include some functions of drugs or biologics are offering innovative treatment options, but can face hurdles in development.

The inherent qualities that characterize a medical device, drug, or biologic product may seem straightforward. These distinctions are becoming increasingly blurred, however, as more new medical products combine qualities of more than one type—such as device with drug, or device with biologic. These combination products are generally complex in nature, and allow the fundamentally different components to work in concert to achieve a greater medical benefit. Such products have far-reaching applications and include cardiovascular devices, tissue implants, orthopedic devices, wound dressings, neurostimulation systems, and organ substitutes.
Until recently, the development of medical products or components has favored selection of materials that would remain biologically inert during contact with a patient's body. Current development efforts often favor material selections that can enhance product performance through the use of materials or coatings that are biologically active. For example, the infection risk associated with a conventional device may be reduced by the addition of a drug-based antimicrobial coating. Combination products also enable the development of previously unavailable treatment options (e.g., certain organ replacement devices are made possible by incorporating active cell tissue that enables the product to replace the function of the human organ).
In the process of seeking innovative solutions to difficult problems in medical treatment through development of combination devices, some companies have inadvertently created new regulatory problems for themselves. Because FDA's expertise and experience in analyzing conventional medical technologies reside in its three centers (Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, and Center for Devices and Radiological Health), a company with experience working with one of the centers does not necessarily know what to expect when another center becomes involved in reviewing a new product.
So long as the product under review allows a clear classification as a device, drug, or biologic, the regulations provide clear guidance on which of the three centers will conduct the evaluation. However, when a product relies on different modes of action, the agency's process for determining the regulatory path to be followed is not always predictable.
CATEGORIZING NEW PRODUCT TECHNOLOGIES
The challenge of establishing jurisdictional authority for products that do not fit neatly into the traditionally defined categories of medical products is not new. The agency issued three separate documents in 1991 to outline procedures for determining how the three centers would cooperate on evaluating data for products incorporating elements that would normally be reviewed by different centers. These agreements define how lead authority is to be assigned to a center in regulating certain categories of devices and provides guidelines on how the centers involved in a given review should interact at specific points in the process.
Charles Durfor, PhD, a senior scientific reviewer at CDRH, says the regulations are quite clear in defining four broad classes of products that will be considered combination products by the agency and will require interaction between centers. The bottom line in classifying a combination product, he says, is considering the primary mode of action. A fundamental factor in defining a product component's function as a medical device—rather than a biologic or drug—is physical or mechanical interaction with the human body. Durfor explains that a product is considered to be a device "if it is intended to affect the function or structure of the body and not dependent on being metabolized." He adds that the language in the act is clear on that issue.
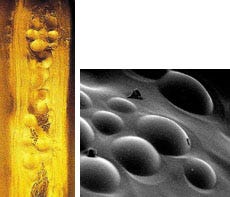 Chromatographic beads were used to simulate the presence of islets in prototype sheets for a bioartificial pancreas (left). Electron micrograph (right) of a sheet made with "faux islets.
Chromatographic beads were used to simulate the presence of islets in prototype sheets for a bioartificial pancreas (left). Electron micrograph (right) of a sheet made with "faux islets.
Combination products become increasingly tricky to classify as researchers discover novel ways to merge diverse technologies. Harry M. Jansen Kraemer Jr., president and CEO of Baxter International Inc. (Deerfield, IL), commented on some of the regulatory and market issues facing medical manufacturers while addressing an audience at the Institute of Medicine workshop "Innovation and Invention in Medical Devices" in February 2000. "A fascinating and important issue arises out of the rapid evolution of technology itself: the fit between new forms of technology and the jurisdiction over those technologies in FDA," he said. Despite the agency having procedures for deciding how to deal with combination products, industry's perception of how well FDA can carry out its task remains somewhat skeptical.
Kraemer also warned that companies attempting to bring unique technologies into the marketplace can be dissuaded from backing more innovative solutions to therapeutic and diagnostic problems if they perceive too many regulatory problems and the governmental agencies do not evolve to keep pace with developments in technology. "For regulatory purposes, what are xenotransplants in which animal organs are genetically engineered to express human surface proteins, thereby suppressing rejection? Are they devices or biologicals? A technology company that is weighing whether or not to push forward in innovative areas such as this must consider the time and risk involved in the hurdles and other procedural matters ahead." Although FDA and the medical product manufacturers both share the same goals, he continued, the healthcare community as a whole should address such issues if U.S. companies are to maintain their lead in introducing significant new technologies to the world market.
ORGAN REPLACEMENT TECHNIQUE USES ESTABLISHED PRINCIPLES
Regardless of the potential regulatory difficulties in developing combination products, some product developers remain enthusiastic about the promises of new and improved technologies to treat human ailments. For example, a bioartificial pancreas under development by Islet Sheet Medical (San Francisco) was conceived as a way to improve the treatment of diabetic patients.
The firm's bioartificial pancreas is not available commercially; however, animal research toward process improvements began in 1998, and preclinical large-animal studies at the University of Cincinnati Medical Center began in February of this year.
 "Dermagraft is a living human dermal replacement used to treat skin that has been injured or destroyed.
"Dermagraft is a living human dermal replacement used to treat skin that has been injured or destroyed.
The implantable device combines living and nonliving components—active islet of Langerhans cells that sense glucose and secrete insulin, and a thin sheet of polymer material. When placed inside the peritoneal cavity, the sheets are intended to replace nonfunctioning islets of Langerhans. Each sheet is several centimeters in diameter, 0.3 mm thick, and contains two to three million living islet cells. Oxygen, glucose, and nutrients diffuse into the sheets to keep the cells alive, while insulin, hormones, and waste diffuse out. The company notes that the technique requires no drug therapy to suppress the immune system, and the sheets may be removed or replaced at any time.
Company president Scott King says he doesn't worry much about dealing with FDA when it comes time to begin the application process since "we have the same safety issues as any implantable." King also expects the regulatory issues to follow the pattern established for devices and biologics similar to the components of the product.
The novel technique is based in part on well-established treatment methods. Says King, "We now have well over 2,000 islet transplants in humans, so it's a well-established medical practice. I am only adding the polymer and packaging it as a device. Given a good safety profile for the transplantation technique, the product should follow established guidelines."
TISSUE PRODUCT TREATS DIABETIC ULCERS
Advanced Tissue Sciences (La Jolla, CA) is another company getting into areas of regulation that straddle the line between devices and biologics. Although a biological component plays a major role in the functioning of the company's Dermagraft wound-dressing product, it was deemed by the agency to function primarily as a device. CDRH took the lead authority for the product, which is still considered to be investigational and incorporates aspects of a device as well as those of a biologic.
Dermagraft consists of human skin tissue grown on a bioabsorbable scaffolding and functions as an implant with live cells to treat burns and certain types of wounds. The device is constructed by seeding dermal fibroblasts onto the three-dimensional scaffold, which is contained in a proprietary, closed bioreactor that simulates the environment of the human body. As the cells grow and divide, they produce collagens, extracellular matrix proteins, and growth factors that are normally found in healthy human dermal tissue.
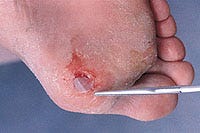 Produced by seeding dermal fibroblasts onto a 3-D scaffold, Dermagraft is implanted into a diabetic foot ulcer to treat damaged skin.
Produced by seeding dermal fibroblasts onto a 3-D scaffold, Dermagraft is implanted into a diabetic foot ulcer to treat damaged skin.
An investigational device exemption (IDE) was approved by FDA in February 2000, allowing clinical trials of Dermagraft to begin for the treatment of diabetic foot ulcers. The firm expects to submit an amendment to its premarket approval application as early as mid-year.
The company suggests that the Dermagraft system could offer a viable treatment option for diabetic foot ulcers, and eventually for other conditions in which the skin is injured or damaged. The dermis of patients with diabetes lacks the collagens, matrix proteins, and growth factors found in normal, healthy skin. Dermagraft has been designed to promote complete wound closure and accelerate healing by addressing these deficiencies. Standard treatments for diabetic foot ulcers include cleaning and bandaging, antibiotics, special shoes that reduce pressure on the foot ulcer, and blood glucose control. Although this approach is sometimes effective, the wounds heal slowly, or may not heal at all. The longer an ulcer remains unhealed, the greater the risk of critical and costly complications—including infection and amputation.
"We have not had difficulty with the regulatory experience," says Gary Gentzkow, MD, the company's executive director of worldwide medical affairs. What is affixed to the wound surface is both an artificial material and a matrix of live cells. "For its purpose of replacing damaged tissue," Gentzkow says, "the decision was that this is a medical device." He explains that, because the product continues to function after implantation by expressing a growth factor, which is a biologic, the classification was not clear-cut. "If a product works mechanically or structurally, and also as a biologic, then [FDA] needs to decide whether it is 54% one and 46% the other."
PREPARING FOR THE REVIEW PROCESS
As with conventional products, combination product manufacturers who are prepared to answer questions about the technology and compliance issues will generally have a better experience with the agency than those that haven't planned for the review process. Elaine Duncan, president of the regulatory and technical consulting firm Paladin Medical Inc. (Stillwater, MN), cautions that manufacturers should be aware of the broader scope of regulations that may affect combination products before they approach FDA for a meeting request.
Though FDA's message to medical manufacturers is that they welcome meetings prior to the filing of formal applications, Duncan advises companies to anticipate the regulatory issues, complete a risk analysis, and go with a prepared submission plan. It may be important to begin with the most familiar experience. "The first option is that you can stay within the branch that you've been working with before on other products. In practice, you don't go straight to the FDA ombudsman's office for a decision on classifying a combination product." she says.
CDRH's Durfor likewise emphasizes that FDA is interested in gathering as much information as possible from a company presenting a new technology for evaluation before getting into questions of regulatory process. Even before a company applies for an IDE, he says, "we encourage companies to come in to talk to us about the product so we can begin to consider the complexity of the technology and even labeling issues." Among the questions that need to be considered in initial meetings between the agency and company are: the regulatory status of each of a product's components; how the product works and what experimental evidence is available about its effectiveness and safety; and some indication of regulatory precedents that might apply.
The FDA Modernization Act established guidelines to help streamline the product approval process, and the agency has adopted a policy of finding a flexible means of operating to develop regulatory solutions to new product classification. In keeping with the philosophy of handling things efficiently within the agency, says Durfor, there is an active spirit of collaboration and consultation between the centers at FDA to share the expertise of each. Regardless of which center hosts the initial meeting with a company, representatives from another center might also be present to consult on different aspects of potential product evaluation issues. "The ultimate goal is to arrive at the most rapid and meaningful review process," he says.
There are situations, however, in which the decision-making process still leaves major questions unanswered. When a device company becomes involved in the manufacture of drugs or biologics, they may become subject to different regulations than they have encountered before, says Duncan. She explains that combination products do not have to constitute a new technology to create new problems for an existing company familiar with manufacturing and marketing requirements in only one FDA center. Even a fairly simple alteration, such as putting a coating on a catheter to enhance its performance, can lead to new regulations that a device manufacturer must address. The question, Duncan concludes, is not whether a company with a combination product can figure out how to work with FDA. "The answer to that is 'Yes, the company can work with the agency,' but the crucial question is, 'Are you really ready for what comes next?'"
NEW TECHNOLOGIES, NEW RISKS
The emergence of new technologies is often accompanied by previously unrecognized risks. For example, the challenge of adding an antimicrobial to the surface of a device has posed unanticipated problems for the makers of this type of combination product. A number of companies succeeded in obtaining premarket clearance for such products only to withdraw them later for various reasons.
Part of the problem with those earlier combination products may be in the understanding of infection control. The medical community has begun to explore whether the overuse of antimicrobials could increase the incidence of opportunistic infections. The question regarding antimicrobial coatings is this: could the coatings be effective for the easy-to-kill microbes, but then leave the surface more vulnerable to colonization by fungus or other, more-resistant pathogens? There appears to be a higher burden of proof in demonstrating the risks and benefits of the altered surface. The need to produce more data demonstrating safety to FDA is clearly greater in these instances.
There is also the question that different divisions or branches within FDA may consider the same coating technology from different perspectives on risks versus benefits. Duncan offers a hypothetical situation involving two manufacturers. One produces a cardiovascular catheter, the other a urological catheter, both of which pose a potential infection risk to patients during prolonged use. Use of the urological catheter often requires multiple catheterizations of the patient, and the risk of infection increases significantly with each subsequent catheter used on the patient. But these two companies may not submit their product application to the same branch at FDA, they may have different FDA reviewers, and each reviewer and each branch may have a different perception of risks and benefits.
Urological catheters sell for less than comparable cardiovascular catheters, and the manufacturer may not be able to afford the cost of an expensive clinical trial to support claims for its urological applications. A coated cardiovascular catheter may sell for more per unit than an uncoated one, so the added cost of the coating or of demonstrating its effectiveness could be absorbed by the higher price. Such differences can influence a manufacturer's decision to add such a coating to the product.
RECOVERING DEVELOPMENT COSTS
Besides the regulatory difficulties of taking a new technology to market, Baxter's Kraemer also identifies another issue that can hold back innovation in medical technology: A company might deem a technology too costly to develop because of potential reimbursement issues. "While payers here in the United States and in other industrialized nations say they are interested in cost-effectiveness, too often it appears to us that their interest is just plain cost cutting. Providers are getting pressure to use cost-saving technology as opposed to cost-effective technology." Increasingly, innovative medical technologies will be combination products that will incur high costs during development and regulatory review. Reimbursement policies, however, do not always take that into account.
The total cost of developing a technology can be affected significantly by the imposition of stricter regulations. So far as FDA is concerned, user fees are imposed by some centers as allowed by legislative and executive mandates. Thus, whether such fees are incurred during product development depends on whether CDER, CBER, or CDRH becomes the lead center in evaluating the new product. Ironically, officers at Advanced Tissue Sciences knew the company's products could easily be considered biologics, so they were prepared to work with CBER rather than CDRH as the lead center and pay the user fees involved.
A larger portion of the costs of manufacturing combination products is usually found in the increased complexity of manufacturing processes and in the added costs of complying with regulations across multiple product categories. The question then becomes whether or not the market will pay a higher price for the products. Modifying devices to add antimicrobial properties has merit in concept, Duncan explains, but concedes that the healthcare marketplace doesn't always respond well to the cost it adds to the basic product.
An example of a hidden extra cost, says Duncan, is the potential change in labeling the addition of a drug component to a device may require. Drugs require shelf-life testing to determine an expiration date, and manufacturers accustomed to open dating may not be prepared for the added expense. Companies compliant with the quality system regulation (formerly GMPs) may be surprised to find they do not comply with the corresponding drug GMPs.
Ultimately the challenge to manufacturers developing combination products may not be so much the increased regulatory burden as the reluctance of the market to accept the cost of developing the technology. Kraemer believes that the industry as a whole needs to consider the value of new technologies—including combination products. "It's hard to argue against a device that works, reduces complications, saves lives, and costs less than the alternatives. But most of the time it's not quite so easy. Chief among these issues is our society's definition of value and the evidence we will accept to prove that value." He adds that the way society defines value in healthcare does not often include a rational assessment of the efforts of industry in developing new technologies.
William Loob is a freelance writer living in Brooklyn, NY.
Opening Illustration by James Schlesinger
Return to the MDDI May table of contents | Return to the MDDI home page
Copyright ©2000 Medical Device & Diagnostic Industry
About the Author(s)
You May Also Like


