




Sign up for the QMED & MD+DI Daily newsletter.
REGULATORY OUTLOOK
+3
20 Min Read
.svg?width=850&auto=webp&quality=95&format=jpg&disable=upscale "Building Quality into Device Clinical Trials, Part 2")
|
Marcarelli |
Within the context of a clinical trial, a sponsor's overall goal is to advance medical technology by ensuring high-quality and robust research while protecting the rights and welfare of study subjects. These responsibilities are outlined in 21 CFR 812, Investigational Device Exemptions (IDEs).1 The regulation lists the following tasks for sponsors:
|
Serge |
• Selecting qualified investigators and providing them with the information needed to conduct the investigation properly.
• Ensuring proper monitoring of the investigation.
• Ensuring that institutional review board (IRB) review and approval are obtained.
• Submitting an IDE application to FDA and ensuring that both FDA and the IRB are informed of significant new information about an investigation.
|
Toth-Allen |
Sponsors should create a well-designed clinical protocol before the study begins. They also need to stress the importance of the informed-consent process and ensure clinical investigator compliance.
|
Harris |
In addition to fulfilling their legal obligations, sponsors can help to expedite the bioresearch monitoring (BIMO) review process. To do so, sponsors must understand what CDRH's Division of Bioresearch Monitoring needs in a submission. Sponsors may also benefit from understanding exactly what will happen during BIMO inspections and from thoroughly preparing staff and clinical investigators for inspection.
|
Immel |
Device sponsors should consider oversight of clinical studies part of their quality system (QS). Device firms already have a QS in place for their manufacturing process, and all of the topics discussed in this article fit nicely within the QS structure under areas such as training, vendor and consultant qualification, design controls, risk management, and corrective and preventive actions.
Selecting Qualified Investigators
Not all doctors are qualified to use a particular device. They may not have the understanding, expertise, or surgical skills necessary to use it. Investigators chosen to participate in a device trial must have the skills and experience necessary to use the device.
Obviously, sponsors want investigators who are committed to the research and who believe it is important to develop new procedures and devices to advance public health. Such commitment helps in recruiting subjects and ensuring confidence in the study outcome.
Often, however, investigators do not realize the amount of time they must invest in conducting research, especially when it comes to documenting that research. Sponsors need to select investigators that have adequate resources, including time, to conduct the study. If a doctor has many patients to see or patients that require more attention than average, the physician may not have time to devote to a study. In addition, the physician should have appropriately trained staff.
Sponsors must also ensure that investigators have all the necessary equipment and ancillary support systems, such as access to required laboratories and diagnostic tests. Finally, the investigator should know all applicable regulations and be familiar with pertinent guidance documents.
Although sponsors should not automatically rule out new doctors who excel in their field, they should ensure that doctors performing their first studies have adequate training in the regulations. Sponsors should consider using a risk management approach to investigator selection and weigh factors that may increase the risk of a low-quality study outcome.
It is important to note that an Investigator's Agreement is a legal contract. CDRH BIMO holds clinical investigators responsible for following the terms of that agreement. Investigators cannot claim lack of knowledge of the regulations if they signed an agreement to follow all applicable regulations.
Finally, investigators need to understand the difference between clinical practice and clinical research. Both clinicians and clinical investigators need to keep patients' needs and safety in mind at all times. A clinician, however, can provide individual patients with new or different treatments, while clinical investigators must follow the protocol. Doctors must also realize the difference between the practice of medicine and a clinical trial when they agree to participate in a study. Lack of protocol adherence may result in a lower-quality submission and could derail a product's entry into the marketplace.
Clinical investigators and all study participants should be reminded that the use of an investigational device cannot be considered a therapy. However, CDRH has expanded access options for investigational devices (e.g., compassionate use and treatment IDEs) that may be used if the treatment under study holds promise and no other satisfactory alternative is available.
Training
Sponsors must ensure that investigators and staff are properly trained on the study protocol. Training should also include the following:
• Study-specific expectations.
• Procedures unique to the device or its use in the study.
• Human factors concerns.
• Regulatory requirements (all applicable regulations).
• The importance of the informed-consent process.
Sponsors should train investigators on any procedures unique to a device or its use, especially regarding any human factors concerns. For example, an instrument that gives a readout may be difficult to understand. There may be important items on the screen that are not obvious and must therefore be pointed out.
Several questions should be answered for any device before a study begins. Are there unique surgical techniques that doctors must learn? What is the learning curve for the device? Does the sponsor need to send a representative to observe the first few uses of the device?
Even experienced investigators should review the precise regulatory requirements for clinical investigators, including their required interactions with both the IRB and with the sponsor. Of course, all training should be documented. Ideally, every clinical site should provide training on good clinical practices on a regular basis to all of its clinical investigators, subinvestigators, and study coordinators, including the principal investigator.
The Compliance Program Guidance Manual on Clinical Investigators details how FDA conducts a clinical investigator inspection.2 Reviewing the guidance with investigators may help sponsors train and prepare them for FDA inspections.
Often, studies are completed well in advance of the FDA inspection. For this reason, sponsors should help each site prepare for inspection by recommending that the staff review all records to familiarize themselves with the study again. They should pull all patient charts, case report forms, source documents, and regulatory binders (containing IRB-approved informed-consent forms, the Investigator's Agreement, adverse events, and so on) and have them readily available during the inspection. They should also dedicate a room for the FDA inspectors for the audit.
Sponsors should provide each site with tips on working with regulatory agency personnel, such as providing an overview of the study to FDA investigators at the beginning of an audit and explaining the usual format of patient charts or case report forms.
Other inspection options include sharing recent FDA warning letters or Form 483s issued to clinical investigators and discussing recent serious clinical cases with them. Any subinvestigators or study coordinators who join the study should also have all necessary, documented training to conduct the study.
Informed Consent
During the training process, sponsors must stress the importance of informed consent. Informed consent should be an ongoing process as new information is learned during the clinical trial. Well-informed subjects who understand the requirements and who know what they have committed to do are less likely to drop out of a study. They are more likely to be compliant with the protocol if they truly understand the details from start to finish.
Sponsors and investigators should give subjects periodic updates. Study subjects may be together in the waiting room quite often, and they may hear something that another subject has experienced as an issue. If sponsors and investigators are continually informing them about the different things that may come up (what they see, what they do not see, and what is important), participants will be less fearful. Such knowledge will also reinforce a subject's commitment to the study. Keeping them informed may also reduce protocol deviations and study dropouts and withdrawals.
Retraining
During a study, the sponsor should provide retraining any time there are significant changes in the device or protocol, or when monitoring reveals problems. Other reasons to provide retraining include the following:
• If follow-up guidance, coaching, and queries do not appear to bring site compliance.
• If there is significant subject noncompliance, which may indicate a need to place a renewed emphasis on the informed-consent process.
• If there are compelling reasons to retain a problematic site.
Clinical investigators and their staffs must understand the consequences of noncompliance with FDA requirements, which include but are not limited to the following:
• An FDA warning letter.
• The study not being accepted in support of a sponsor's claims of safety and effectiveness in the research and marketing permit.
• Sponsor termination of the IDE.
• Initiation of disqualification proceedings against (or entry into a consent agreement with) the clinical investigator.
Make sure that all training and retraining is documented. The original record should be kept at the site, but the sponsor should also retain a copy. If sponsors find problems, they should reconnect with clinical investigators. Sponsors may need to help a site manage its informed-consent process too. Site personnel may not be getting the full picture to the subjects at the beginning, so patients may not really understand their commitment. In other situations, sponsors will want to provide retraining. The longer any significant noncompliance continues, the more study data are put in jeopardy. According to 21 CFR 812, sponsors (not FDA) are responsible for ensuring clinical investigator compliance.1
Monitoring
|
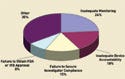
Figure 1. (click to enlarge) Common sponsor inspection deficiencies reported in 2005. |
Monitoring the data life cycle is probably the most important thing a sponsor can do to keep a study on track and to maintain quality. In 2005, 24% of FDA inspections found problems with monitoring, and inadequate monitoring continues to be the top deficiency cited in FDA inspections of sponsors (see Figure 1).3 Building quality into the research process can prevent numerous queries and late database problems. During monitoring, sponsors can also provide training for new or noncompliant study staff and for any changes to the protocol.
It is critical to ensure data quality at each stage of the data life cycle. For example, during a study, monitoring should be done early and frequently enough to meet the needs of the specific study.
Of course, because every study is different, the term early depends on the situation. A good rule of thumb is to begin monitoring after a few subjects have been treated to ensure that the sites understand and are following protocol. Catching mistakes early could save an entire study.
Monitoring should follow well-developed standard operating procedures (SOPs). 21 CFR Part 812 requires written SOPs for significant-risk device studies (those that require FDA approval to start).1 Critical areas to review during site monitoring visits include the following:
• Human-subject protection (informed consent, IRB review and approval of study documents, and periodic review of the study by the IRB).
• Compliance with the investigator agreement, investigational plan, and regulations.
• Source document verification.
• Case report forms (CRFs) for currency, completeness, and accuracy.
• Device accountability for currency, completeness, and accuracy.
Sponsors should ensure that all patients sign an informed-consent form before being enrolled in the trial and that the form (and the protocol) were reviewed and approved by an IRB. They also need to make sure that the most current version of the informed-consent form is used, that all subjects were given sufficient time to consider their participation in the study, and that the consent form contains all required elements.
Including research oversight in a device firm's QS audit procedures is a good way to confirm quality and ensure that the study is functioning within acceptable limits. If nonconformance occurs, this information can then be sent to the clinical management review team for proper evaluation and resolution.
Ensuring Compliance
Every sponsor should have a predetermined strategy to ensure clinical investigator compliance. Tasks such as expeditious reviews of monitoring and auditing reports, taking immediate action to correct noncompliance, and, if necessary, being able to halt shipments of investigational devices, fall to the sponsor. If all else fails, the offending site's participation in a study may be terminated, and sponsors should report that termination to CDRH's Office of Device Evaluation (ODE).
|
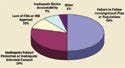
Figure 2. (click to enlarge) Clinical investigator inspection deficiencies reported in 2005. |
The regulations require sponsors to bring noncompliant investigators into compliance (see Figure 2 for common investigator difficulties). CDRH BIMO recommends that sponsors sensitize clinical investigators to their compliance plan up front and include these options in the investigator agreements.
Most IDEs are approved for a limited number of subjects. If many subjects come from a site with problems and if data from those subjects are not going to be usable to support a premarket approval (PMA) or 510(k), sponsors may want to notify their CDRH reviewer.
|
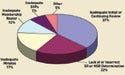
Figure 3. (click to enlarge) IRB inspection deficiencies reported in 2005. SR and NSR refer to significant risk and nonsignificant risk, respectively. |
Finally, if a sponsor needs to close a site because of noncompliance, a sponsor will still need to follow up with the study subjects. At that point, it is important to find staff at the site who are willing to complete the follow-up. If subjects were terminated from the study because the site was terminated and the IRB refuses to give the sponsor access to data, the sponsor needs to have someone willing and able to oversee the patients. Whenever possible, choose clinical investigators that are likely to complete the necessary follow-up with the patients (see Figure 3 for common IRB deficiencies).
Developing The Protocol
Input from potential clinical investigators, CDRH Scientific Review Groups, other regulatory bodies (e.g., NIH or CMS), or industry consultants should be sought before finalizing the study protocol. Inclusion and exclusion criteria must be appropriate and not restrictive. The timing of procedures should be clinically appropriate and any testing, both initially and at follow-up visits, should satisfy critical study endpoints. CDRH generally advises sponsors to consult with study design experts and with experts in the appropriate field of medicine when designing the protocol. Adherence to protocol will be carefully assessed during FDA inspections of the clinical investigator sites.
Preventing Protocol Violations. FDA recommends that sponsors include the definition of a violation in the protocol. Deviations occur when something is done differently from how it should have been done, and when something is not done that should have been done. During the study, a thorough examination of the monitoring reports can uncover evidence of recurring violations. Analyze the violations to determine whether they are site specific or are found across the study.
Common site-specific violations include omission of initial or follow-up testing, inclusion of a specific group of inappropriate subjects, a missing or inaccurate set of data elements, and frequent subject noncompliance. For site-specific problems, the root causes may include personnel or training issues.
If problems occur across sites, a discussion should be initiated with the clinical investigators to determine the root causes and whether the study is salvageable. That decision may require a discussion with the CDRH reviewing division. Consider putting new enrollment on hold until your decision is made. Questions to ask include the following:
• Where is the protocol deviation occurring?
• Is it a recurring deviation?
• Is the deviation occurring during initial or final testing?
• Is the error happening because the test is not considered clinically necessary?
• Are the inclusion and exclusion criteria correct?
• Are certain data elements consistently missing or inaccurate?
• Are the required data correct?
• Is subject noncompliance high?
After an investigation is finished, sponsors may have to start again, or at least expand their population. Sponsors may even have to choose a different endpoint or use different tests. In other words, they will need to redesign the protocol in retrospect.
Reviewing the Data Set. Errors happen, but their effect can be minimized with careful planning. When deciding what data to collect, consider the following:
• Collect only data critical to the safety and effectiveness endpoints.
• Meet all applicable regulatory requirements and guidance recommendations.
• Capture data using checklists limited to numbers, results, or a few descriptive words.
• Avoid duplication of data in a patient's chart or on the case report forms.
• Talk with the CDRH reviewing division when appropriate.
It is reasonable to expect that a certain percentage of errors will occur in any given data set. If there is no applicable guidance document, discuss the plans with the appropriate FDA review division. Ask which types of data reviewers will want for the device being investigated. Such discussions are particularly important for new types of devices.
Recordkeeping Tips. Clinical investigators or study coordinators should initial and date CRFs when recording answers or when documenting test results. They should note whether the information is from the subject or the subject's diary. They should also authenticate who is recording the information. Some information may come from questions asked by the clinical coordinator.
Training records should include a list of the attendees, a sign-in sheet, or other written documentation of the site training. If there was a problem, is there documented evidence of retraining? Is there evidence of suitable training for all investigators (including the principal investigator and any subinvestigators) and all study coordinators who worked on the study?
When using subject questionnaires, instead of putting information directly on the CRFs, keep the completed questionnaire in the patient's medical record or chart. If a procedure or process needs to be done at the hospital, make a copy of the record to keep in the subject's file or chart and maintain access to the original document at the hospital. Any form sent to clinical investigator sites becomes the source document for their entries. The more pieces of paper in a file, the more chance there is of making an error. So try to minimize the number of copies.
For electronic CRFs, FDA requires that any system used must provide accurate and complete information regarding the conduct of the study. One way a firm can do this is through the use of a computer-generated audit trail. The FDA investigator will look at the data and review the audit trail, the actual data entered, and any changes to the entered information. FDA recommends validating electronic CRF software before rolling it out to the sites. Make sure that data are routinely backed up. Having an accurate, complete, and readable back-up copy of completed CRFs at each clinical investigator site is crucial.4,5
Device Accountability. Sponsors must have adequate device accountability at the clinical site and at their own facilities. Sites with missing records (including who received investigational devices, where they went, and whether they were returned) raise a regulatory red flag.
Expediting the BIMO Review Process
|
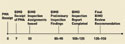
Figure 4. (click to enlarge) BIMO review milestones (in calendar days). |
PMA Submissions. All sponsors want an expeditious review of their applications. PMA applications undergo multiple simultaneous reviews within FDA (see Figure 4), including a parallel review by CDRH BIMO and Scientific Review Divisions.6 Sponsors should prepare their submissions so that all the necessary data are readily accessible to both the reviewers and the inspection team. When a submission is made, the sponsors and clinical investigators should be ready for inspection.
It may be a good idea for the sponsor to inform clinical investigators when it submits a marketing application. FDA wants to perform early BIMO inspections to give timely feedback to the review team. Problems identified late in the review mean wasted time for the sponsor and for FDA.
Sidebar: |
BIMO Inspection Process. Before inspection, CDRH BIMO reviews pertinent volumes of the PMA to gather the necessary information for inspection. BIMO tells the sponsor when information is missing and, most frequently, the data tabulations or the complete clinical investigator information is what is missing. A sponsor usually has this information readily available, so CDRH recommends including it within the submission in a Microsoft-compatible format.
BIMO reviewers consult agency databases for the inspectional histories of the sponsor and investigator sites. They consult with the lead FDA reviewer about his or her inspection site preferences and draft an inspection assignment with pertinent attachments. BIMO inspection assignments typically carry a 90-day due date from CDRH receipt of the PMA submission to completion of the inspection.
BIMO reviewers enter the assignment into the agency database and distribute attachments to district BIMO coordinators. They consult with FDA field investigators as necessary and provide interim reports to CDRH scientific reviewers on inspectional findings and their relevance to the review process. Later, they review the completed inspection report and draft postinspectional correspondence. They also provide the final BIMO review memo to the CDRH scientific reviewers.
In general, inspections are study specific and issued for a multitude of reasons. Such reasons include complaint follow-up, early intervention (e.g., novel technology, vulnerable population, etc.), in-house PMA or 510(k), or surveillance (e.g., IRB or nonclinical lab).
Sometimes reviewers have specific concerns about a study. For example, one site's results may look different from all the other sites (either better or worse). It may be a matter of numbers, such as too few subjects at one site or too many at another, or it may be that the investigator has a history of noncompliance. The concerns might also relate to the quality of a sponsor's submission.
Conclusion
In 2005, inadequate monitoring and inadequate device accountability were the most frequent citations issued to sponsors by FDA. Other problems included investigators who did not follow the investigational plan or the regulations, inadequate subject protection or inadequate informed consent, and protocol adherence.
As many as 37% of the IRBs have inadequate initial or continuing review of the study. Other problems include lack of or incorrect significant and nonsignificant risk determination, and inadequate minutes. The IRB is one part of the three-pronged oversight of the studies. Oversight responsibilities are shared among the sponsor, the clinical investigator, and the IRB.
CDRH BIMO usually reserves some of its inspectional resources to look at studies while they are in progress (i.e., early intervention). This helps to ensure that systems are in place to protect human subjects and that data collected during actual research are of high quality. CDRH BIMO expects that the early intervention program will result in higher-quality submissions and enhance the availability of new medical technologies.
Acknowledgements
The contents of this article are the authors' views. They do not necessarily reflect the views or policies of FDA or its staff. FDA will not be bound by any information contained in this article. This article is based on a three-hour electronic conference presented on November 22, 2005, by members of FDA's Division of Bioresearch Monitoring, Office of Compliance, CDRH. The conference was cosponsored by the Clinical Device Group and FDLI. Special thanks go to Nancy Stark of the Clinical Device Group for providing a CD copy of the conference for use in preparing this article.
Michael Marcarelli, Marian Serge, Jean Toth-Allen, and Cynthia Harris all work in the Division of Bioresearch Monitoring in the Office of Compliance at CDRH. Barbara Immel is president of Immel Resources LLC (Petaluma, CA) and can be contacted at [email protected].
References
1. Code of Federal Regulations, 21 CFR Part 812.
2. “Compliance Program Guidance Manual 7348.811, Clinical Investigators” (Rockville, MD: FDA, 1997); available from Internet: www.fda.gov/ora/ftparea/compliance/48_811.pdf.
3. Michael E. Marcarelli et al., “Building Quality into Device Clinical Trials, Part 1,” Medical Device & Diagnostic Industry 28, no. 7 (2006): 60–70.
4. Code of Federal Regulations, 21 CFR Part 11.
5. “Guidance for Industry, Part 11: Electronic Records, Electronic Signatures: Scope and Application” (Rockville, MD: FDA 2003); available from Internet: www.fda.gov/cder/guidance/5667fnl.pdf.
6. “Draft Guidance for Industry and FDA Staff: The Review and Inspection of Premarket Approval Applications under the Bioresearch Monitoring Program” (Rockville, MD: FDA, 2006); available from Internet: www.fda.gov/cdrh/comp/guidance/1602.pdf.
Copyright ©2006 Medical Device & Diagnostic Industry
About the Author(s)
You May Also Like


