Cardiovascular
FDA approved Boston Scientific's Farapulse system in January for pulsed field ablation treatment of atrial fibrillation.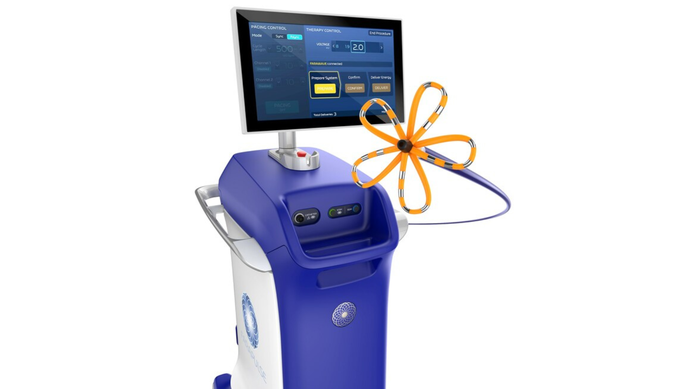
CardiovascularBoston Scientific's Farapulse Is a ShowstopperBoston Scientific's Farapulse Is a Showstopper
Electrophysiologists are giving the new pulsed field ablation system a warm welcome.












.png?width=300&auto=webp&quality=80&disable=upscale "Medtronic's Aurura EV ICD (extravascular implantable cardioverter-defibrillator) is designed with the lead placed outside of the heart and veins")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")


.png?width=300&auto=webp&quality=80&disable=upscale "BD Vascular Covered Stent")

.png?width=300&auto=webp&quality=80&disable=upscale "Agent Drug-Coated Balloon")
.png?width=300&auto=webp&quality=80&disable=upscale "Freudenberg Medical")

.png?width=300&auto=webp&quality=80&disable=upscale "Abbott Laboratories TriClip system")



.png?width=300&auto=webp&quality=80&disable=upscale "anatomical heart")

.png?width=300&auto=webp&quality=80&disable=upscale "Bare Temporary Spur Stent System")
.png?width=300&auto=webp&quality=80&disable=upscale "Micra VR")
.png?width=300&auto=webp&quality=80&disable=upscale "Farapulse")




.png?width=300&auto=webp&quality=80&disable=upscale "Embolic device")
Editors' Choice


Sign up for the QMED & MD+DI Daily newsletter.