The medtech news you need. In one minute or less.
May 22, 2024
Apr 19, 2024
Apr 18, 2024
From Innovation to Commercialization: Navigating the Surgical Robotics Market
Medtech in a Minute: US Authorities Throw the Book at Philips, and More
Porex Partners With UK-based Diagnostics Startup
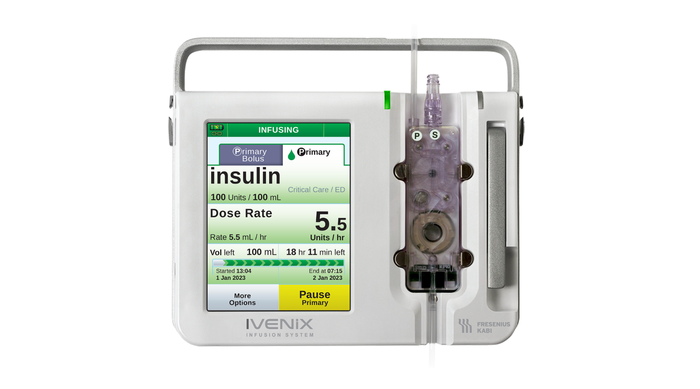
Fresenius Kabi USA Infusion Pump Software Recall Declared Class I





















.png?width=300&auto=webp&quality=80&disable=upscale "plastic syringe")


.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "EU MDR")
.png?width=300&auto=webp&quality=80&disable=upscale "Axonics R20 Rechargeable Sacral Neuromodulation System")


.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Elon Musk")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA Recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Shane Farritor")

.png?width=300&auto=webp&quality=80&disable=upscale "Regulatory Pathway")

.png?width=300&auto=webp&quality=80&disable=upscale "FDA Safety Communication")
.png?width=300&auto=webp&quality=80&disable=upscale "European Union Council")




