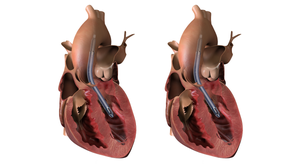
Abbott HeartMate 3 heart pump (left ventricular assist device) designed to help the heart pump blood to the rest of the body.
QA/QC14 Deaths Connected to Latest Abbott HeartMate Recall14 Deaths Connected to Latest Abbott HeartMate Recall
Long-term buildup of biomaterials can obstruct the device, making it less effective at helping the heart pump blood.


















.png?width=300&auto=webp&quality=80&disable=upscale "plastic syringe")


.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "EU MDR")
.png?width=300&auto=webp&quality=80&disable=upscale "Axonics R20 Rechargeable Sacral Neuromodulation System")


.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Elon Musk")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA Recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Shane Farritor")

.png?width=300&auto=webp&quality=80&disable=upscale "Regulatory Pathway")

.png?width=300&auto=webp&quality=80&disable=upscale "FDA Safety Communication")
.png?width=300&auto=webp&quality=80&disable=upscale "European Union Council")




.png?width=300&auto=webp&quality=80&disable=upscale "verification")
.png?width=300&auto=webp&quality=80&disable=upscale "Medical Device Recall")
Editors' Choice


.png?width=300&auto=webp&quality=80&disable=upscale)
Sponsored Content
May 23, 2024
Sign up for the QMED & MD+DI Daily newsletter.