LumiSystem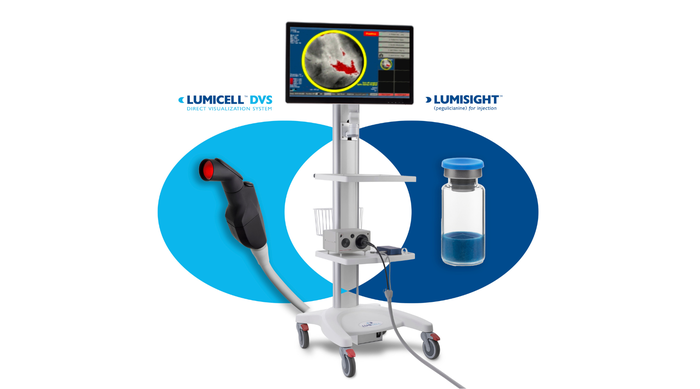
Medical Device RegulationsBreast Cancer Detection Drug-Device System Nabs FDA ApprovalBreast Cancer Detection Drug-Device System Nabs FDA Approval
The LumiSystem enables surgeons to scan the breast cavity post-lumpectomy in real time, detect, and resect residual cancer that otherwise could have been missed.





















.png?width=300&auto=webp&quality=80&disable=upscale "plastic syringe")


.svg?width=300&auto=webp&quality=80&disable=upscale "thePACKout 2024 Conference Agenda")
.png?width=300&auto=webp&quality=80&disable=upscale "EU MDR")
.png?width=300&auto=webp&quality=80&disable=upscale "Axonics R20 Rechargeable Sacral Neuromodulation System")


.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Elon Musk")
.png?width=300&auto=webp&quality=80&disable=upscale "FDA Recall")
.png?width=300&auto=webp&quality=80&disable=upscale "Shane Farritor")

.png?width=300&auto=webp&quality=80&disable=upscale "Regulatory Pathway")

.png?width=300&auto=webp&quality=80&disable=upscale "FDA Safety Communication")
.png?width=300&auto=webp&quality=80&disable=upscale "European Union Council")
Editors' Choice

Sign up for the QMED & MD+DI Daily newsletter.