News
thumbnail
CardiovascularNew Medical Devices & Partnerships Could Break Barriers & Lead to Better Heart Failure CareEmerging Medical Devices & Partnerships Could Power Better Heart Failure Care
New technologies like wireless patch electrocardiograms and cellular-connected scales aim to change remote heart failure care, potentially leading to earlier detection and more efficient management.
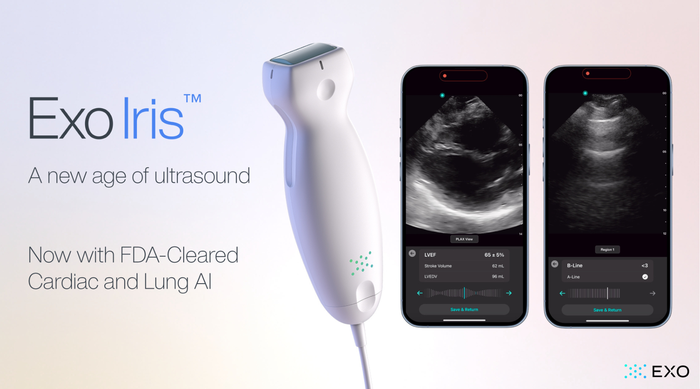

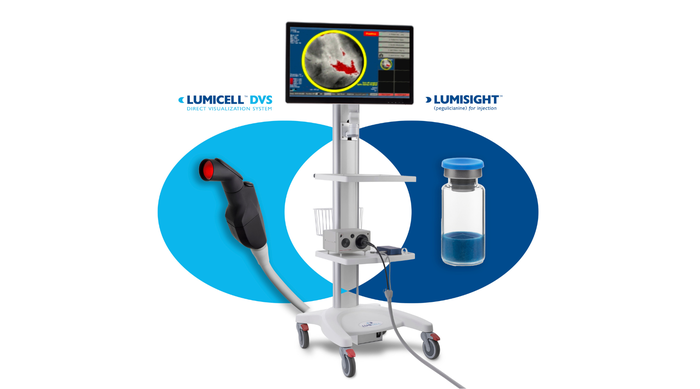










 device being deployed to repair a leaky tricuspid valve")


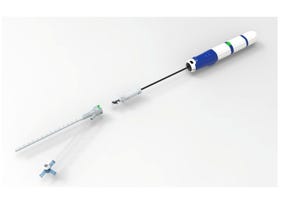







 designed to help the heart pump blood to the rest of the body.")













Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.