thumbnail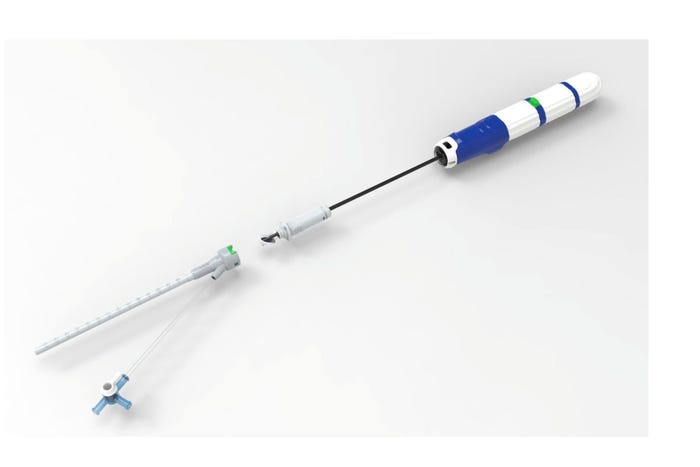
CardiovascularVivasure Opens up about Securing a Top Spot in Vessel Closure MarketVivasure Makes an Impact in the Growing Vessel Closure Market
Recent approvals in the TAVR and TMVR markets are providing significant opportunities for companies like Vivasure Medical. The Galway, Ireland-based company recently said the first large-bore venous patient was treated with the PerQseal Elite vascular closure system in a clinical trial.




.png?width=700&auto=webp&quality=80&disable=upscale)


















.png?width=300&auto=webp&quality=80&disable=upscale "NeuroStar")

.png?width=300&auto=webp&quality=80&disable=upscale "Siemens Healthineers")


.png?width=300&auto=webp&quality=80&disable=upscale "Medtronic's Aurura EV ICD (extravascular implantable cardioverter-defibrillator) is designed with the lead placed outside of the heart and veins")

.png?width=300&auto=webp&quality=80&disable=upscale "Laura Perryman convicted")

.png?width=300&auto=webp&quality=80&disable=upscale "FDA recall")

Editors' Choice



Sign up for the QMED & MD+DI Daily newsletter.